
miR-7-5p抑制胰腺癌细胞放疗后加速再增殖的体外实验研究
2020-06-12 03:02叶志强郭贵龙陈涵斌吕奇原谷甸娜
肝胆胰外科杂志 2020年5期
叶志强,郭贵龙,陈涵斌,吕奇原,谷甸娜
(温州医科大学附属第一医院,浙江 温州 325000,1.外科,2.放化疗科)
胰腺癌是目前已知恶性程度最高的肿瘤之一,五年生存率只有5%左右,其病死率基本接近肿瘤的发生率[1]。其早期诊断困难、易于侵袭转移,现在仍缺乏有效的诊治方法。目前,手术虽是根治胰腺癌的首要手段,但绝大部分胰腺癌患者在被诊断时已局部进展或者发生远处转移,失去了手术治疗的机会,因此只能接受放化疗。然而,胰腺癌对放化疗并不敏感,表现为明显的治疗抵抗。其中肿瘤细胞放疗后再增殖是导致放疗抵抗,引起治疗失败的一个重要原因[2]。因此,如能抑制放疗后再增殖将能一定程度减少肿瘤细胞的放疗抵抗。放疗的关键靶点是细胞的DNA。放射线引起DNA损伤后,机体会启动损伤应答通路,这种通路的激活将会阻滞细胞周期,可以让受损肿瘤细胞有足够的时间来进行自我修复,继而产生放疗抵抗,而加速再增殖与肿瘤细胞的DNA损伤修复能力有关。近年来报道,DNA损伤诱导转录子4(DNA damage inducible transcript 4,DDIT4)可参与应激、DNA损伤修复和细胞凋亡等多种生命过程,并在乳腺癌、前列腺癌和胃癌等肿瘤中表达增高[3]。那DDIT4有否通过影响DNA损伤修复进而影响放疗后再增殖呢?另外,肿瘤细胞在接受放疗后会出现大量细胞死亡,细胞死亡后会释放多种细胞内容物,包括DNA、RNA和蛋白等。已有研究表明miRNA可通过影响DNA损伤修复、细胞周期检查点、凋亡、信号转导、肿瘤组织微环境等因素调控肿瘤辐射敏感性[4]。据报道miR-7可通过抑制EGFR-PI3K-AKT信号通路改善肺癌、乳腺癌、头颈部肿瘤及胶质瘤细胞的放疗敏感性[5],而且我们前期研究发现miR-7-5p可抑制胰腺癌细胞增殖促进凋亡[6],那么miR-7-5p可否抑制放疗后再增殖呢?因此,本研究通过建立的胰腺癌细胞放疗后再增殖模型,研究濒死细胞释放的miR-7-5p对放疗后再增殖效应的影响,并从分子水平探讨相关机制。
1 材料和方法
1.1 细胞培养
人胰腺癌细胞系PANC-1细胞购于中国科学院典型培养物保藏委员会细胞库,使用DMEM培养基进行培养,置于5% CO2、37 ℃的培养箱中培养。根据细胞倍增时间决定传代时间,用0.25%胰酶进行消化传代,选择对数生长期的细胞作为实验对象。
1.2 慢病毒包装
在6孔板中接种3×105个293细胞,加入2 mL DMEM完全培养基,置于37 ℃,5% CO2培养箱中培养24 h。细胞汇合度达到80%~90%时,按照Lipofectamin 2000脂质体说明书,将三质粒系统(pLEXGFP-luc2、psPAX2和pMD2.G)共转染至293细胞中,转染48 h后,收集培养上清液。4 ℃ 3 000 g离心10 min,去除细胞碎片,然后将上清用0.45 μm的PVDF滤膜过滤,-80 ℃分装保存。
1.3 慢病毒感染和稳定表达细胞株的构建
感染前24 h将细胞以每孔1×105密度接种于6孔板中含有10% FBS的DMEM培养基中进行培养,待细胞贴壁后长至50%左右进行慢病毒感染,在6孔板中加入已滤病毒液,于5% CO2的37 ℃培养箱中培养,病毒感染48 h后使用荧光显微镜进行观察,感染细胞出现绿色荧光,使用添加嘌呤霉素的10%FBS DMEM培养基进行筛选,获得稳定表达荧光素酶-绿色荧光蛋白(Fluc-GFP)的PANC-1细胞,记作PANC-1-LUC细胞。后续通过检测PANC-1-LUC细胞的荧光素酶活性,反映细胞增殖的情况[7]。
1.4 PANC-1细胞放疗后再增殖模型构建及分组
待PANC-1细胞贴壁生长进入对数分裂期后,将其铺于实验所需的孔板中,次日使用线性加速器给予单次10 Gy放射线处理细胞。接着将放疗后的肿瘤细胞当天消化计数,接种于24孔板中,每组设置3个复孔。24 h后,将稳定表达Fluc-GFP的PANC-1-LUC细胞接种于24孔板上的Transwell培养室(0.4 μm孔径)内进行共培养,根据处理方法不同分为3组:PANC-1-LUC+PANC-1(10 Gy)组、PANC-1-LUC+PANC-1(0 Gy)组及PANC-1-LUC组。当研究受辐照细胞对PANC-1-LUC细胞辐照后损伤修复等影响时,给予PANC-1-LUC细胞单次2 Gy照射,分为2组:PANC-1-LUC(2 Gy)+PANC-1(10 Gy)组与PANC-1-LUC(2 Gy)+PANC-1(0 Gy)组。根据实验指定时间点,向24孔板中加入荧光素酶底物,利用小动物活体成像仪对细胞进行化学发光成像。
1.5 Real-time PCR检测miRNA-7-5P的表达水平
采用miRNeasy Serum/Plasma Advanced Kit(Qiagen)提取上清液中的miRNA,按照PrimeScriptTMRT reagent kit(TakaRa)逆转录试剂盒说明书将RNA逆转录合成cDNA。在QuantStudio 6 Flex实时荧光定量PCR仪中,使用SYBR®Premix Ex TaqTM试剂盒,量化miRNA-7-5P的表达水平。
1.6 流式细胞仪检测细胞周期及细胞凋亡
用胰酶消化后收集细胞,在预冷的75%乙醇固定过夜,加入PI/RNase常温染色30 min,PBS洗涤,流式细胞仪检测细胞周期,使用Modfit软件进行数据分析。凋亡采用PE标记的Annexin-V和7-AAD凋亡试剂盒,各加5 μL,室温孵育30 min,再用PBS洗涤,用流式细胞仪检测凋亡率。
1.7 双荧光素酶法验证miR-7-5p靶基因为DDIT4
利用Targetscan软件对miR-7-5p的靶向基因进行预测筛选,预测了与辐射损伤修复相关基因DDIT4为其下游靶基因。设计合成扩增靶基因DDIT4-3'UTR序列的引物如下:Forward 5'-TCAGAGCTCA CTTCAACCTGAGGGGGCCGACA-3’;Reverse 5'-G TGGCTAGCTGTTTTAACAAACATGTTTATTAG-3'。并将合成的DDIT4-3'UTR、野生型(WT)序列及突变型(MT)序列进行退火,对DDIT4-3'UTR、野生型及突变型用SacI/NheI双酶切,构建pmirGLO(Promega)双荧光报告质粒,所得重组质粒均经测序鉴定。将miR-7-5p mimics与对照空载体、克隆有3'UTR序列或对应的WT、MT序列的报告质粒分别共转染进入PANC-1细胞,转染48 h后,以海肾荧光素酶活性作为内参,检测萤火虫荧光素酶活性/海肾荧光素酶活性的相对值,统计检测结果。
1.8 Western blotting检测γH2A.X与DDIT4蛋白
取对数生长期的细胞,使用加有蛋白酶抑制剂的蛋白裂解液RIPA收集蛋白,冰上裂解40 min后离心15 min,收集上清即为蛋白样品。采用SDS-PAGE电泳法对提取的蛋白进行电泳分离,然后印迹于PVDF膜上。膜在5%脱脂牛奶中封闭1 h,加入一抗在4 ℃下孵育过夜,在室温下,将膜与HRP标记的二抗孵育2 h,洗膜后显影。
1.9 统计学分析
所有数据应用SPSS 19.0软件进行统计分析,计量资料以(±s)表示,计量资料两组间均数比较采用Student'st检验,多组间均数分析采用单因素方差分析,两两比较采用LSD-t检验,P<0.05为差异有统计学意义。
2 结果
2.1 受辐照PANC-1(10 Gy)细胞对PANC-1-LUC细胞加速再增殖及DNA损伤修复的影响
受辐照PANC-1(10 Gy)细胞在放疗后第3天明显抑制了PANC-1-LUC细胞的生长,且这个抑制效应在第5天才消除,此后与受辐照PANC-1(10 Gy)细胞共培养的PANC-1-LUC细胞进入快速的增殖阶段,表现为明显的再增殖效应(图1)。与此同时,我们观察到受辐照PANC-1(10 Gy)细胞能明显促进PANC-1-LUC(2 Gy)细胞放疗后γH2A.X的消退,提示受辐照PANC-1(10 Gy)细胞促进了X线照射后细胞的DNA损伤修复(图2)。
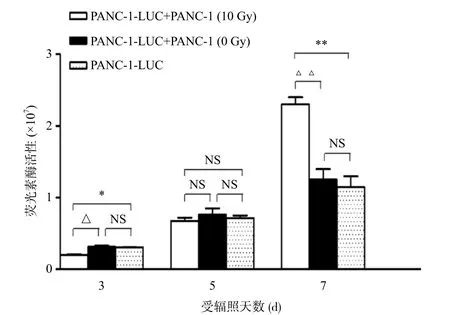
图1 受辐照PANC-1(10 Gy)细胞促进PANC-1-LUC胰腺癌细胞加速再增殖
2.2 受辐照PANC-1(10 Gy)细胞对PANC-1-LUC(2 Gy)细胞凋亡和细胞周期的影响
与PANC-1-LUC(2 Gy)+PANC-1(0 Gy)组相比,PANC-1-LUC(2 Gy)+PANC-1(10 Gy)组不仅抑制了存活PANC-1-LUC(2 Gy)细胞的凋亡(图3),此外还增加了PANC-1-LUC(2 Gy)细胞G0/G1期的阻滞(图4)。

图2 受辐照PANC-1(10 Gy)细胞促进PANC-1-LUC(2 Gy)细胞放疗后DNA损伤修复
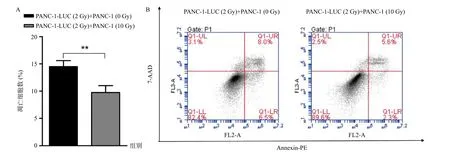
图3 与受辐照PANC-1(10 Gy)细胞共培养抑制放疗后PANC-1-LUC(2 Gy)细胞凋亡
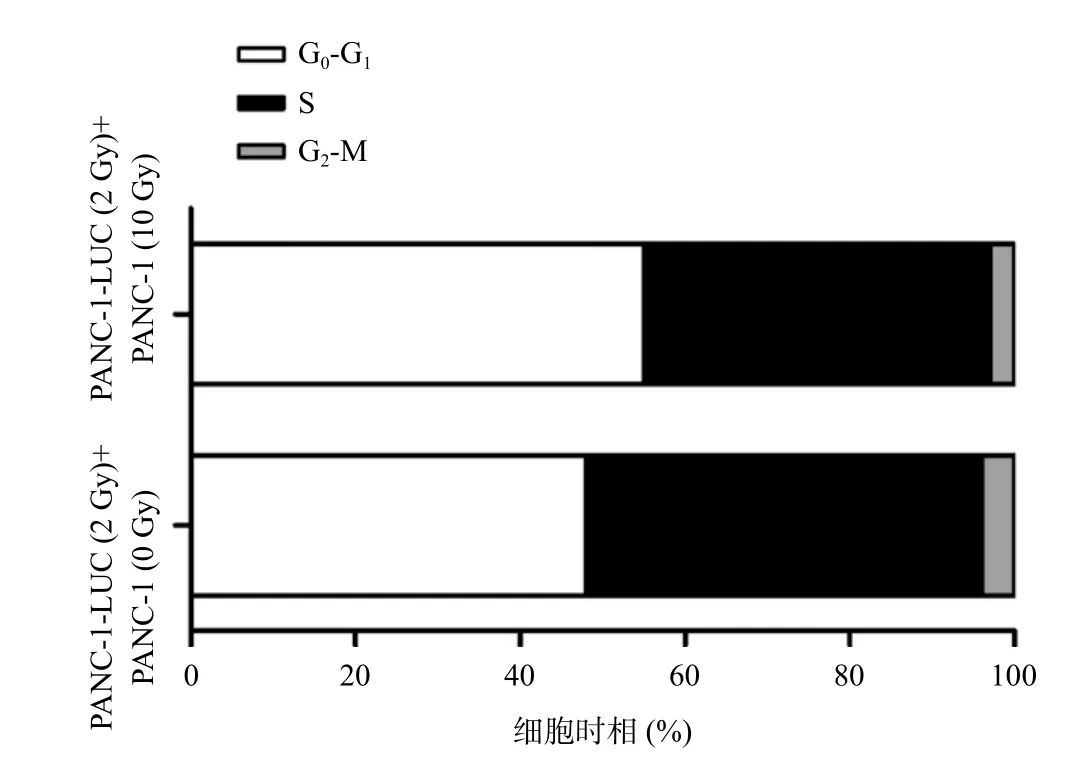
图4 与受辐照PANC-1(10 Gy)细胞共培养增加PANC-1-LUC(2 Gy)细胞的G0/G1周期阻滞
2.3 miR-7-5p表达上调可抑制PANC-1-LUC(2 Gy)细胞放疗后加速再增殖
我们利用RT-PCR方法检测受辐照PANC-1(10 Gy)细胞上清液中miR-7-5p的表达情况,结果提示miR-7-5p在受辐照PANC-1(10 Gy)细胞的上清中明显表达下调(t=16.9,P<0.01)。有研究报道miR-7-5p具有抑制肿瘤细胞生长的能力,而我们也在放疗处理后的细胞上清中检测到miR-7-5p释放减少。因此,我们猜想是否因放疗抑制的miR-7-5p促进了残余肿瘤细胞增殖,从而参与了肿瘤细胞再增殖。为了证实我们的猜想,我们往再增殖模型中加入miR-7-5p mimics,观察是否对肿瘤再增殖有影响。成像结果显示,PANC-1-LUC(2 Gy)+PANC-1(10 Gy)+miR-7-5p mimics组的荧光素酶活性明显低于PANC-1-LUC(2 Gy)+PANC-1(10 Gy)组(P<0.01) (图5)。因此,我们认为提高miR-7-5p水平能够显著抑制PANC-1细胞放疗后再增殖。
2.4 miR-7-5p在胰腺癌细胞中通过靶向DDIT4促进放疗后加速再增殖
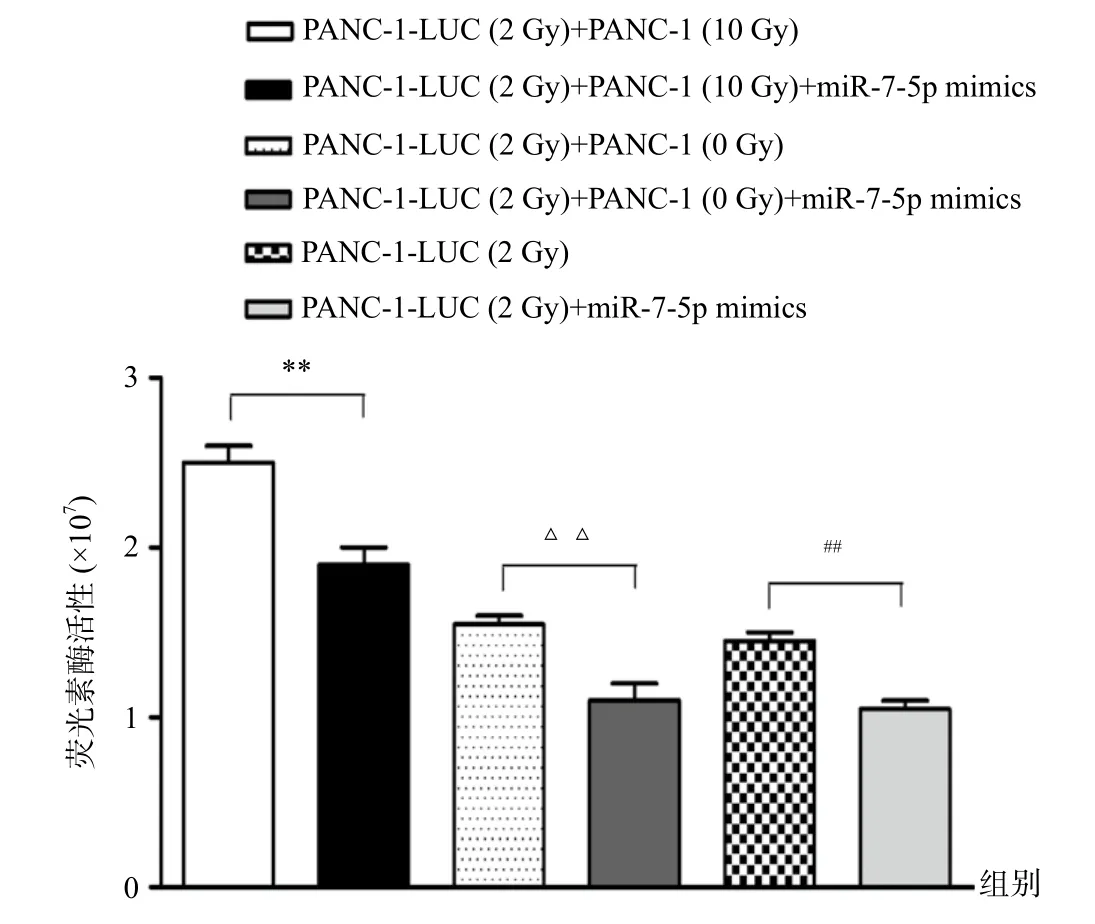
图5 上调miR-7-5p水平抑制胰腺癌细胞放疗后加速再增殖
为了探究miR-7-5p调控胰腺癌细胞放疗后加速再增殖的分子机制,我们利用生物信息学方法,获得miR-7-5p的潜在靶基因,并根据Targetscan筛选出与DNA损伤修复相关的DDIT4基因,我们用双荧光素酶活性实验进行验证。在DDIT4-3'-UTR和野生型WT组中,miR-7-5p mimics的加入使胰腺癌PANC-1细胞中荧光值显著降低(3'-UTR组:t=12.6,P<0.01;WT组:t=10.8,P<0.01)。在DDIT4突变型MT组中,PANC-1细胞荧光值未见显著变化(t=1.01,P>0.05),证明胰腺癌细胞中通过miR-7-5p靶向DDIT4基因发挥调控作用(图6A~B)。进一步,我们通过West-ern blotting证实在转染miR-7-5p mimics的PANC-1细胞中,DDIT4蛋白明显抑制,反之,转染miR-7-5p inhibitor可上调PANC-1细胞DDIT4蛋白的表达(图6C)。所以,miR-7-5p在胰腺癌细胞中靶向调控DDIT4分子。

图6 miR-7-5p在胰腺癌细胞中靶向调控DDIT4分子
接着,我们进一步利用Western blotting检测放疗后共培养时DDIT4分子及凋亡信号的表达水平。与PANC-1-LUC(2 Gy)+PBS组相比,PANC-1-LUC(2 Gy)+PANC-1(10 Gy)组DDIT4蛋白表达明显升高,并且抑制了凋亡信号转导。反之,在培养上清液中加入miR-7-5p mimics则可抑制DDIT4信号,诱导细胞凋亡(图7A~B)。
3 讨论
几十年来尽管临床上一直强调应重视肿瘤加速再增殖现象,但至今对于肿瘤放疗加速再增殖的机制尚未完全清楚。放疗过程中,由于受到组织增殖动力学及细胞周期的影响,肿瘤细胞受到照射后会在放疗后期出现增殖加速。研究证实,细胞周期、DNA损伤修复机制和蛋白调节等多种信号通路可能与加速再增殖有关[8]。研究发现,经放疗后的濒死细胞可以通过SHH-Wnt信号[9],活化caspase-3/7-PKCδ信号[10],以及上调Sox211等刺激胰腺癌存活细胞加速再增殖。我们既往研究也曾报道,放疗后濒死细胞可通过miR-193a调控TGF-β2/TGF-βRIII信号通路促进胰腺癌细胞放疗后再增殖[7]。
本研究通过经荧光素酶标记的胰腺癌细胞PANC-1-LUC建立了胰腺癌细胞放疗后加速再增殖模型,我们发现接受单次辐照PANC-1(10 Gy)细胞,在放疗后3天明显抑制了存活的PANC-1-LUC胰腺癌细胞的生长,且这个抑制效应在第5天才得以解除,之后进入快速的增殖阶段,荧光强度明显增加,这提示加速再增殖现象的存在。进一步地,检测比较经辐照与未辐照处理后的细胞上清液中miR-7-5p的表达情况,结果显示受辐照PANC-1(10 Gy)细胞上清液中miR-7-5p明显下调,提示miR-7-5p在放疗后加速再增殖中起了重要作用。既往研究报道,miR-7在多种实体肿瘤中如肝癌[12]、肺癌[13]、乳腺癌[14]、胃癌[15]等多种肿瘤生物学过程中起到重要的抑制作用,并且可改善乳腺癌细胞、肺癌细胞、喉鳞状细胞癌及神经胶质瘤细胞的放疗敏感性[16-17]。而我们先前在胰腺癌中的研究,也证实miR-7-5p可抑制细胞的生长和增殖、阻滞细胞周期,提示miR-7-5p在胰腺癌中亦起到抑癌作用[6],那miR-7-5p是怎么影响放疗后加速再增殖的呢?本研究将受辐照PANC-1(10 Gy)细胞的上清液中补充miR-7-5p mimics与PANC-1-LUC(2 Gy)细胞共培养,相比于PANC-1-LUC(2 Gy)组,PANC-1-LUC(2 Gy)+PANC-1(10 Gy)+miR-7-5p mimics组细胞荧光强度明显减少,同时发现存活细胞中miR-7-5p的靶基因DDIT4信号蛋白也明显受抑制。
电离辐射等会对细胞造成一定程度的DNA损伤,细胞内某些机制将促使发生细胞周期阻滞,使细胞有足够时间完成损伤修复或DNA复制,从而维持基因组的稳定,但这正是细胞放射抵抗的重要原因[18]。而DDIT4是一种DNA损伤诱导的转录因子,既往研究报道,在胃癌中,DDIT4可通过影响p53和丝裂原活化蛋白激酶(MAPK)信号通路[19],促进胃癌细胞的增殖和胃癌的发生。在膀胱癌中,DDIT4促进癌细胞增殖减少凋亡[20]。此外,DDIT4可介导肿瘤自噬促进肿瘤存活,并与卵巢癌患者预后呈负相关[21-22]。在本研究中,放疗后濒死细胞上清液中miR-7-5p表达减少,使存活胰腺癌细胞中DDIT4显著上调,通过调控生长阻滞和细胞凋亡来保持基因组完整性,促进了放疗后加速再增殖。反之,当上清液中miR-7-5p上调,存活胰腺癌细胞中DDIT4表达下调,则抑制DNA损伤修复,抑制胰腺癌细胞放疗后再增殖。
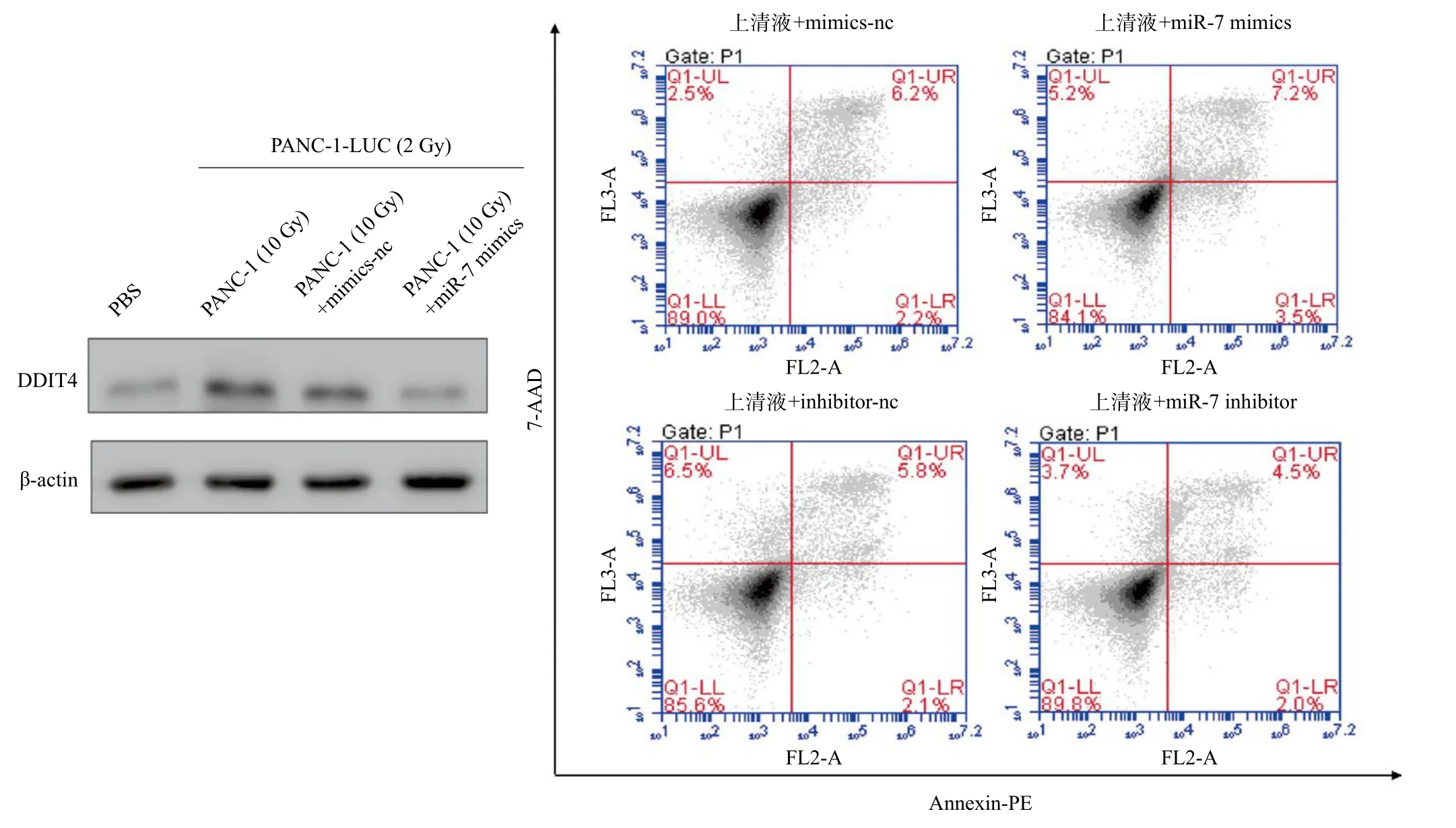
图7 miR-7-5p通过抑制DDIT4促进放疗后PANC-1-LUC(2 Gy)细胞凋亡
综上所述,本研究发现放疗后胰腺癌细胞可通过下调miR-7-5p促进DDIT4表达,从而抑制细胞凋亡及调控细胞周期再分布来加速再增殖,表明提高miR-7-5p的水平能够提高胰腺癌细胞的放疗敏感性,这将为改善胰腺癌放疗抵抗提供了一种新的思路和方法。
猜你喜欢
核科学与工程(2022年3期)2022-10-18
天津医科大学学报(2021年4期)2021-08-21
中日友好医院学报(2021年1期)2021-04-14
宁夏医学杂志(2020年3期)2021-01-21
山东医药(2020年9期)2020-05-20
现代矿业(2018年9期)2018-10-16
天然产物研究与开发(2018年4期)2018-05-07
船海工程(2015年3期)2015-10-21
中国医药导报(2015年27期)2015-02-28
中国医疗美容(2015年5期)2015-02-03
