
长白山北部西伯利亚狍局域种群间的基因流
2020-08-03 06:36田玉苗盛清宇袁立成姜广顺
野生动物学报 2020年3期
田玉苗 盛清宇 袁立成 姜广顺
(国家林业和草原局猫科动物研究中心,东北林业大学野生动物与自然保护地学院,哈尔滨,150040)
广义的基因流是指,导致一个群体向另一个群体内产生基因交换的所有机制的通称。而基因迁移,即基因流,是群体间产生基因交换或遗传物质交换的某种表现形式[1]。因此,在群体水平或个体水平层面中,在群体或个体消亡或重建过程中所涉及的一切基因的“运动”或遗传物质的动态交换过程,都可以统称为基因流。基因流通常被认为是重要的群体分化驱动力,来维持分化或正在分化群体间的稳定性,使得群体适应环境。基因流是影响群体内部和群体之间遗传变异程度的重要因素,也是产生进化的一个重要因素。
狍(Capreolusspp.)是鹿科(Cervidae)中分布最广泛的动物之一,包括2个种,即体型相对较小的欧洲狍(Capreoluscapreolus)以及体型相对较大的西伯利亚狍(Capreoluspygargus)[2-3]。西伯利亚狍的主要分布区域包括整个亚洲大陆的古北界[2]及东欧的部分地区[4]。在中国,狍主要分布在北部各省份,按种类分为东北亚种、西北亚种和天山亚种。在东北虎(Pantheratigrisaltaica)、东北豹(Pantherapardusorientalis)分布区域内,也广泛分布,是东北豹的主要猎物(占食物构成的66%)以及东北虎的猎物之一(占食物构成的9%)[5]。因此,研究狍种群间的基因流,对于探究猎物种群的有效恢复,对东北虎、东北豹种群的保护有着重要的意义。
目前,有相当部分的关于欧洲狍种群基因流的研究,如Coulon等[6]研究表明,欧洲狍的基因流受景观连通性的影响。此外,Baker等[7]使用线粒体 DNA 和微卫星标记的研究方法,比较了英国原有的狍种群和引入种群之间的遗传多样性差异以及种群混合和扩散。Hepenstrick等[8]研究表明,在瑞士全国重要的野生动物走廊内,高速公路围栏是狍基因流动的主要障碍。而关于西伯利亚狍的种群间基因流的研究,不管在国外还是国内都相对较少。且大多数关于西伯利亚狍的研究,多集中于西伯利亚狍种群遗传多样性研究,如Lee等[9]对亚洲 10 个地区西伯利亚狍的12个微卫星位点的变异进行了研究,分析了各个种群的遗传多样性水平,揭示了西伯利亚狍存在的3个不同地理种群。仅有盛清宇[10]使用10个微卫星位点研究西伯利亚狍的偏性扩散,其结果表明西伯利亚狍的扩散机制为偏雌扩散。但其仅研究西伯利亚狍种群的扩散机制,未对种群间基因流进行研究。总的来说,目前国内利用微卫星标记分析西伯利亚狍种群间基因流的研究基本上处于空白状态。鉴于此,本研究试图利用9个微卫星位点,结合荧光标记PCR技术,使用贝叶斯方法评估地理种群间的基因流,并探讨长白山北部西伯利亚狍局域种群间基因流对群体遗传分化的影响,以期为管理、制定保护政策提供遗传学基础。
1 材料与方法
1.1 样品采集
2015—2017年,根据西伯利亚狍种群在长白山北部地区地理分布范围,选择5个地理采样区域(图1),包括张广材岭(ZGCL)、珲春东北虎国家级自然保护区(HC)、汪清国家级自然保护区(WQ)、穆棱林业局和天桥岭林业局(MT),东宁林业局(DN)。主要为冬季样线调查或跟踪调查时采集样本。共收集279份粪便样本。为了保持样品的质量,在提取DNA之前,粪便样品被-80 ℃保存。
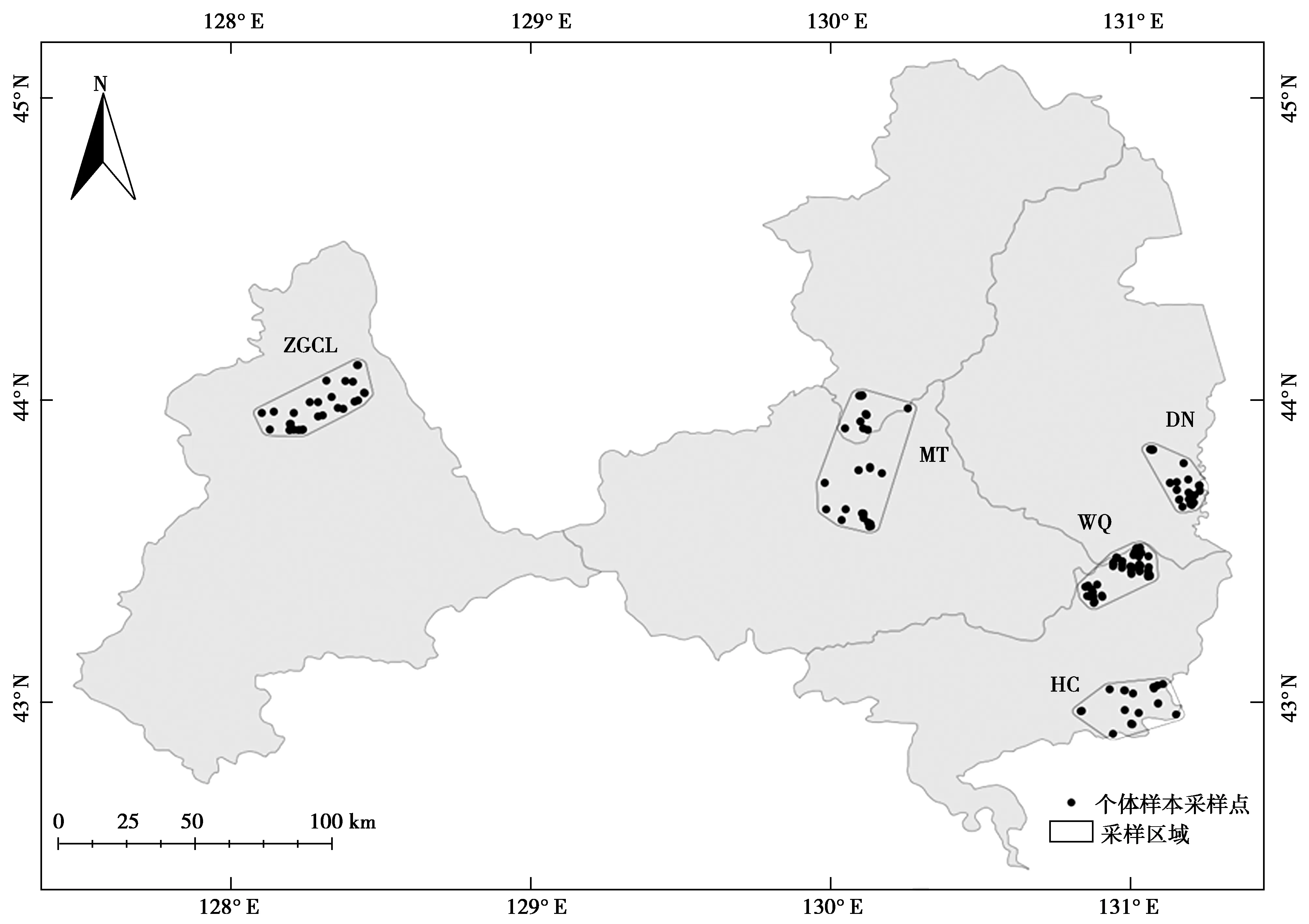
图1 研究区域地理位置示意图和粪便个体采样位置Fig.1 A geographical map of the study area and the location of individual fecal samples
1.2 粪便 DNA 提取
使用QIAamp DNA粪便微型试剂盒从刮下的粪便表面提取总基因组DNA。 总共提取了279个DNA样品。用于后续实验步骤。袁巍[11]推荐的物种特异性引物用于物种鉴定。
Pta-CbF(5′-CTAATCTCATCAATCCTAATC-3′).
Pta-CbR(5′-TTGGTATGAGTACTAGAATA-3′).
在1.0%琼脂糖凝胶中鉴定出222 bp的细胞色素b序列,并在琼脂糖凝胶上和UV透射照明器上可视化。我们在20 μL体系中使用KOD FX Neo DNA聚合酶(TOYOBD),并添加1 μL DNA,退火温度为51 ℃。
1.3 微卫星位点及引物选择
微卫星是由2—5个核苷酸串联的重复序列构成,由于其多态性高,信息量大,实验步骤少且简单,实验结果稳定,DNA 需求量少等优点,广泛应用于遗传学分析。本研究选取了9个微卫星位点并合成引物,进行荧光染料标记。本研究所使用的微卫星位点均为2碱基重复,引物序列信息见表 1。
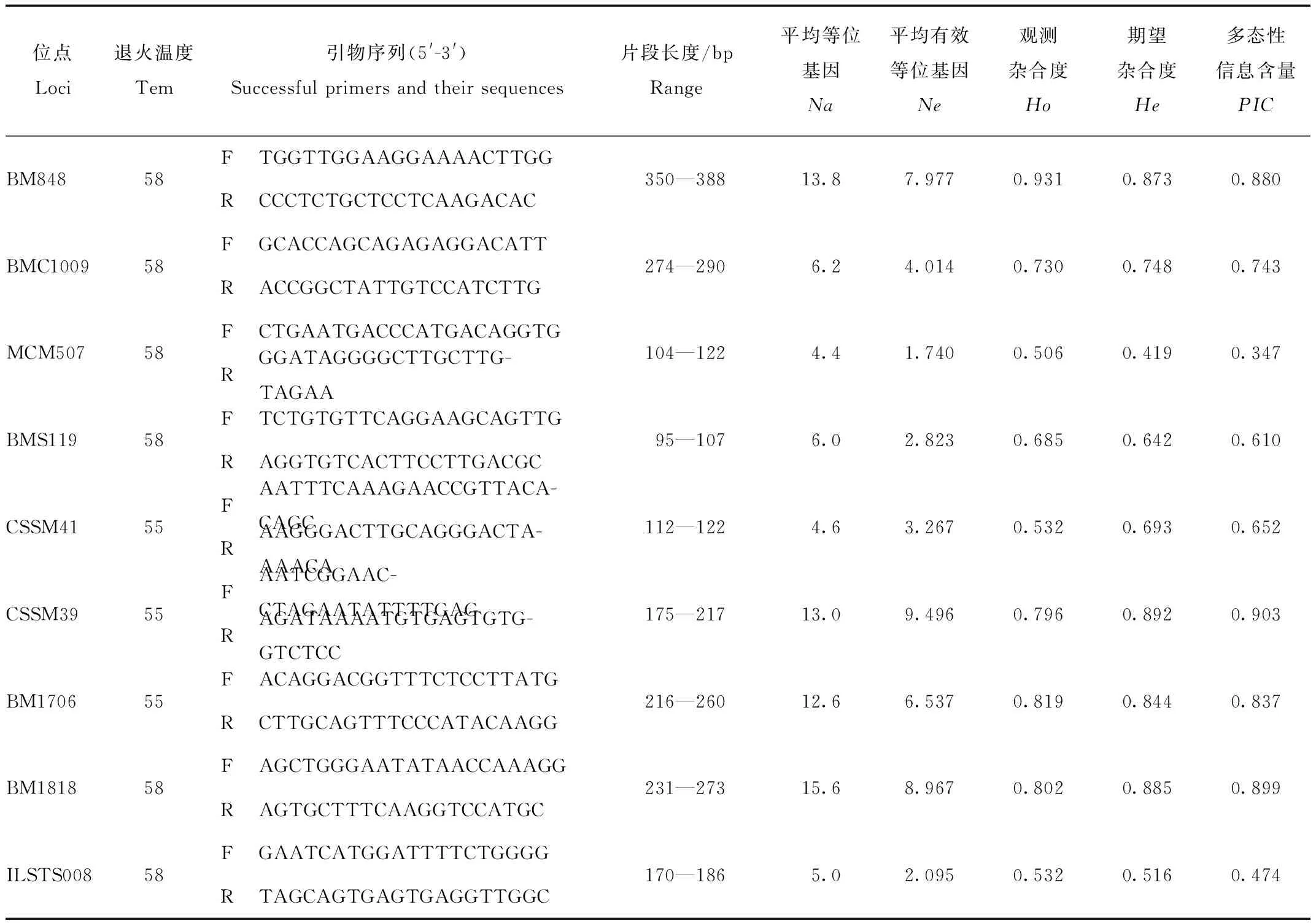
表1 9个微卫星位点信息,遗传多样性参数和多态性信息含量(PIC)、平均等位基因(Na)、平均有效等位基因(Ne)、观测杂合度(Ho)和期望杂合度(He)Tab.1 9 microsatellite loci,genetic diversity parameters and polymorphism information content(PIC),mean allele(Na),mean effective allele(Ne),observed heterozygosity(Ho)and expected heterozygosity (He)
1.4 PCR扩增
PCR扩增的反应体系为:10 μL 10 × PCR Buffer,4 μL dNTP Mixture(2.5 mmol/L),各0.75 μL的上/下游引物 (10 mmol/L),0.3 μL TOYOBO高保真酶,1.0 μL的模板DNA,使用ddH2O补足体系。PCR循环参数为:94 ℃预变性2 min;94 ℃变性20 s,55—58 ℃退火1 min,68 ℃延伸1 min,35个循环;68 ℃延伸10 min。4 ℃保存。扩增完毕后,取5 μL PCR扩增产物在含有核酸染料的 1% 琼脂糖凝胶中,以 120 V 的电压电泳30 min,以确定PCR扩增是否成功。每个样本进行PCR扩增3—5次,以确保实验结果的准确性。使用ABI3730X1 测序仪对所有微卫星位点荧光标记下的PCR扩增产物进行基因型分型。
1.5 统计分析
使用Micro-checker[12]来检验无效等位基因和等位基因缺失。使用Excel Microsatellite Toolkit v 3.1.1[13]进行个体鉴定。使用Cervus v.3.0.7[14]计算位点多态信息含量(PIC)。使用GenAlEx v.6.1[15]计算每个位点和每个地理种群的等位基因数(Na),有效等位基因数(Ne),观察(Ho)和期望(He)杂合度。另外,通过GenAlEx v.6.1测试了每个位点上每个种群的Hardy-Weinberg平衡(HWE)。使用GENEPOP v.4.0.11[16-17]测试位点的连锁不平衡(LD)。利用 FSTAT v.2.9.3[18-19]软件计算两两种群之间的FST。
BayesAss 1.3[20]允许居群偏离哈温平衡,允许居群大小不同和迁移不对称。应用MCMC分析和Bayesian方法,通过确定居群近交系数和连锁不平衡,计算当代基因流,BayesAss 能估测个体迁移历史的后验概率(posterior probability),因此该方法可以用来估测5个地理种群间的扩散情况和邻接种群间的当代基因流。使用多组种子数(seed number)和迭代次数,选择对数似然值(log-likelihood values)最大时的不作数迭代次数(buin-in length),将参数的变化值设为总迭代次数的40%—60%。本实验最终使用3×106的迭代次数,106的不作数迭代次数,设置取样频次(sample frequency)为2 000,以此获得稳定一致的后验概率。
2 结果
2.1 遗传多样性
利用软件 MicroChecker 2.2.3 对本研究使用的9个微卫星位点进行等位基因检测时,3 种估算方法(Oosterhout、Chakraborty 和 Brookfield法)均未探测到等位基因缺失或无效等位基因的存在。使用Microsatellite Toolkit进行个体鉴定。结果表明,HC有21个个体,ZGCL有32个个体,WQ有82个个体,DN有37个个体,MT有43个个体。从279个粪便样本中成功鉴定出215个个体(图1)。
每个位点和每个种群共有45个哈迪-温伯格平衡试验,21个显著偏离哈迪-温伯格平衡(表2)。在位点间仅检测出少量位点在个别种群中可能存在连锁现象,未见任何位点在3个以上种群中与其他位点连锁,因此这9对微卫星引物可以用于后续的分析研究。
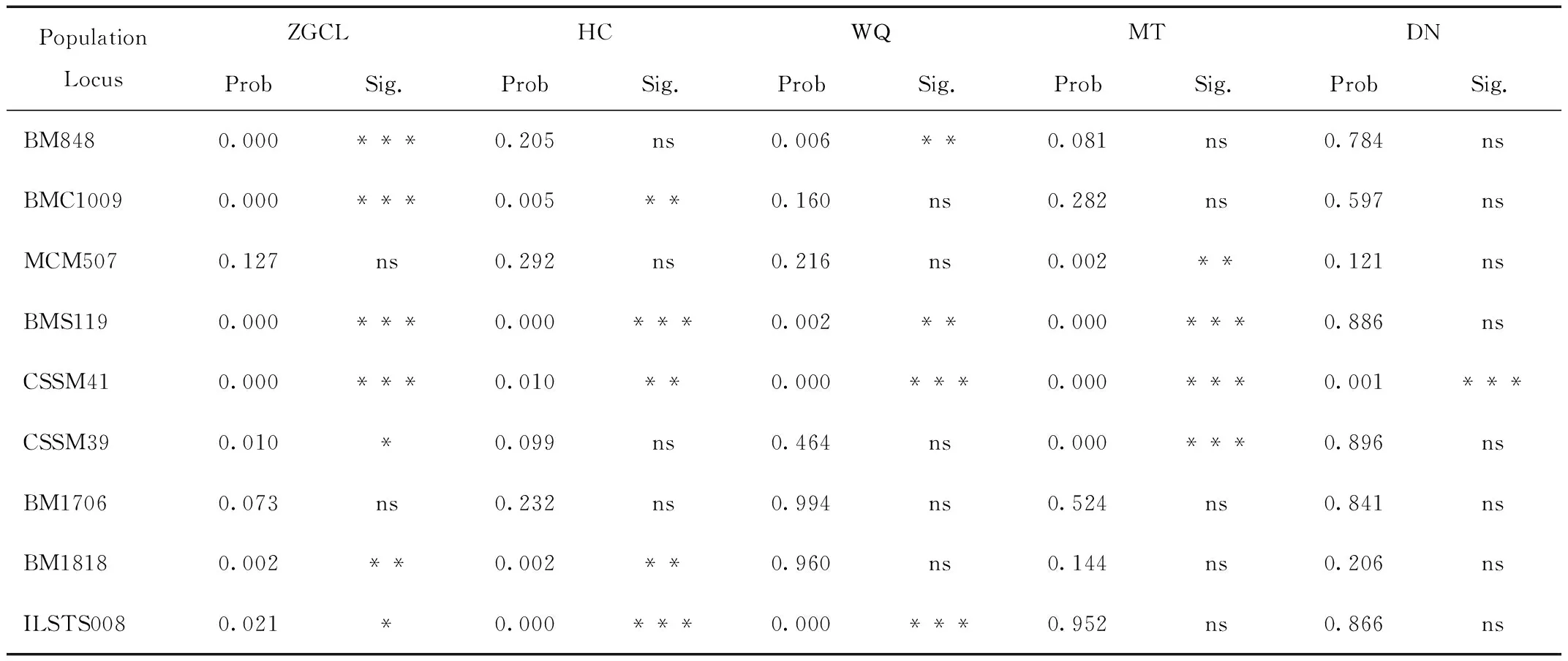
表2 各种群 9 个微卫星位点的 Hardy-Weinberg平衡卡方检验的 P 值及显著性Tab.2 P value and significance of Hardy-Weinberg equilibrium chi-square test for 9 microsatellite loci in various groups
9个位点为中-高多态性,等位基因范围为4.4—15.6个(表1)。5个群体中等位基因的平均数量为8.556—10.111。5个群体中等位基因的平均数量为8.556—10.111。由于WQ群体中个体数量最多,等位基因的数量会因个体数量的不同而有很大的差异。5个群体的观测杂合度为0.693—0.730,期望杂合度为0.714—0.732(表3)。FST评估表明种群分化程度较低(FST范围为-0.001 1—0.018 0,表4)。总体上,90%(9/10)的两两群体分化值与0差异显著(P<0.05)。
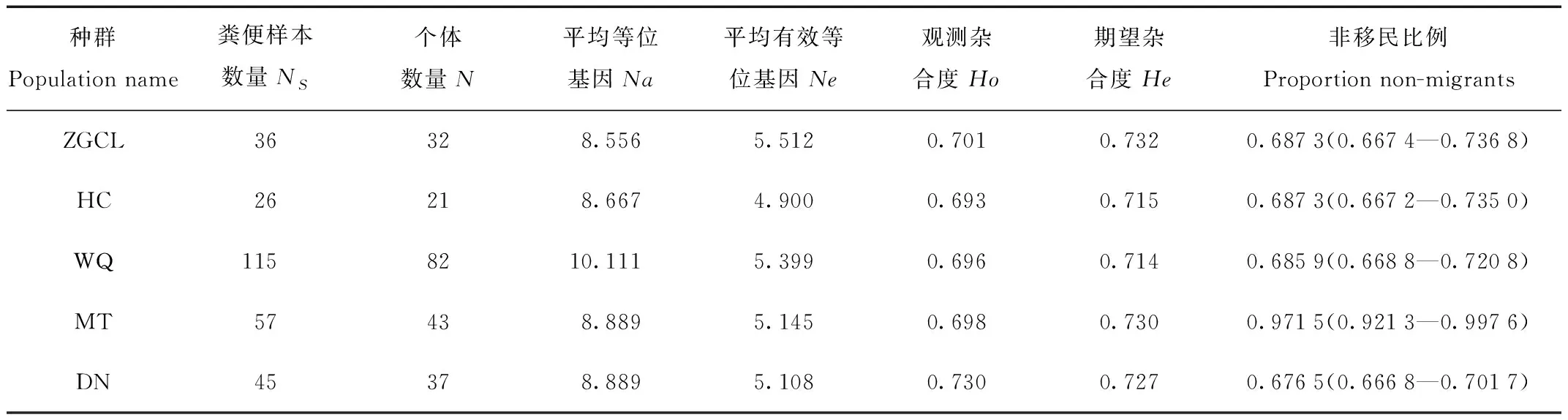
表3 西伯利亚狍种群遗传多样性参数以及使用BayesAss1.3程序计算了5个地理种群在近1—3代之间的非移民比例(proportion non-migrants,95% CI)。粪便样本数量(NS)、个体数量(N)、平均等位基因(Na)、平均有效等位基因(Ne)、观测杂合度(Ho)和期望杂合度(He)Tab.3 The parameters of genetic diversity of the Siberian roe deer population and the BayesAss 1.3 program were used to calculate the proportion of non-migrants(proportion non-migrants,95% CI)of the five geographical populations between the last and the third generation.Number of fecal samples(NS),number of individuals(N),mean allele(Na),mean effective allele(Ne),observed heterozygosity(Ho)and expected heterozygosity(He)
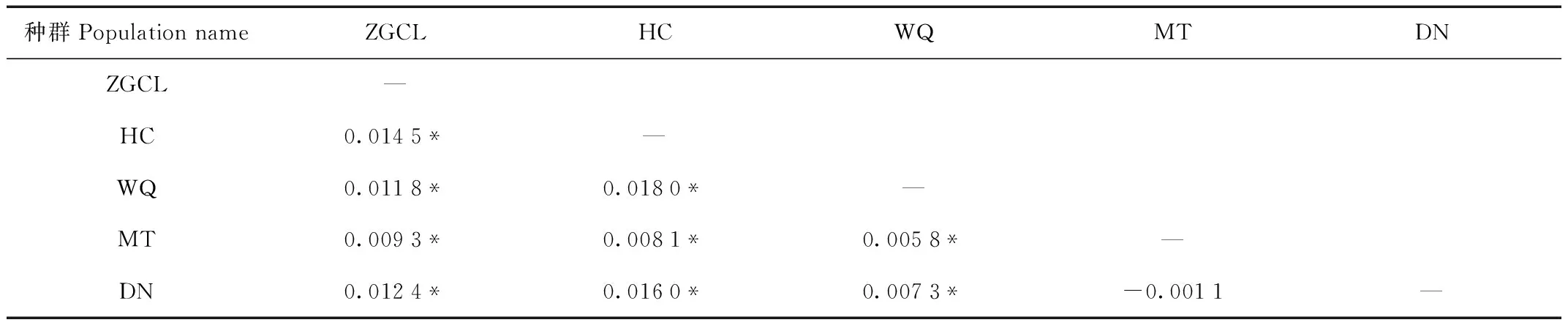
表4 种群间遗传分化系数FSTTab.4 Population comparisons with FST values
2.2 基因流
使用 BayesAss 1.3计算的基于贝叶斯法的种群间定向基因流表明,5个地理种群间出现不对称基因流(表5,图2),基因流从MT种群流向其他种群,其他种群间基因流动对称,同时MT种群有很高比例的“非移民”个体(97.15%),表明MT是分散的源种群[21](表3)。其余各种群间也存在相当程度的基因交流,如WQ到ZGCL(2.46%),ZGCL到HC(1.01%),WQ到HC(1.23%),DN到HC(1.51%)等,说明群体间存在较为广泛的不对称的基因流。
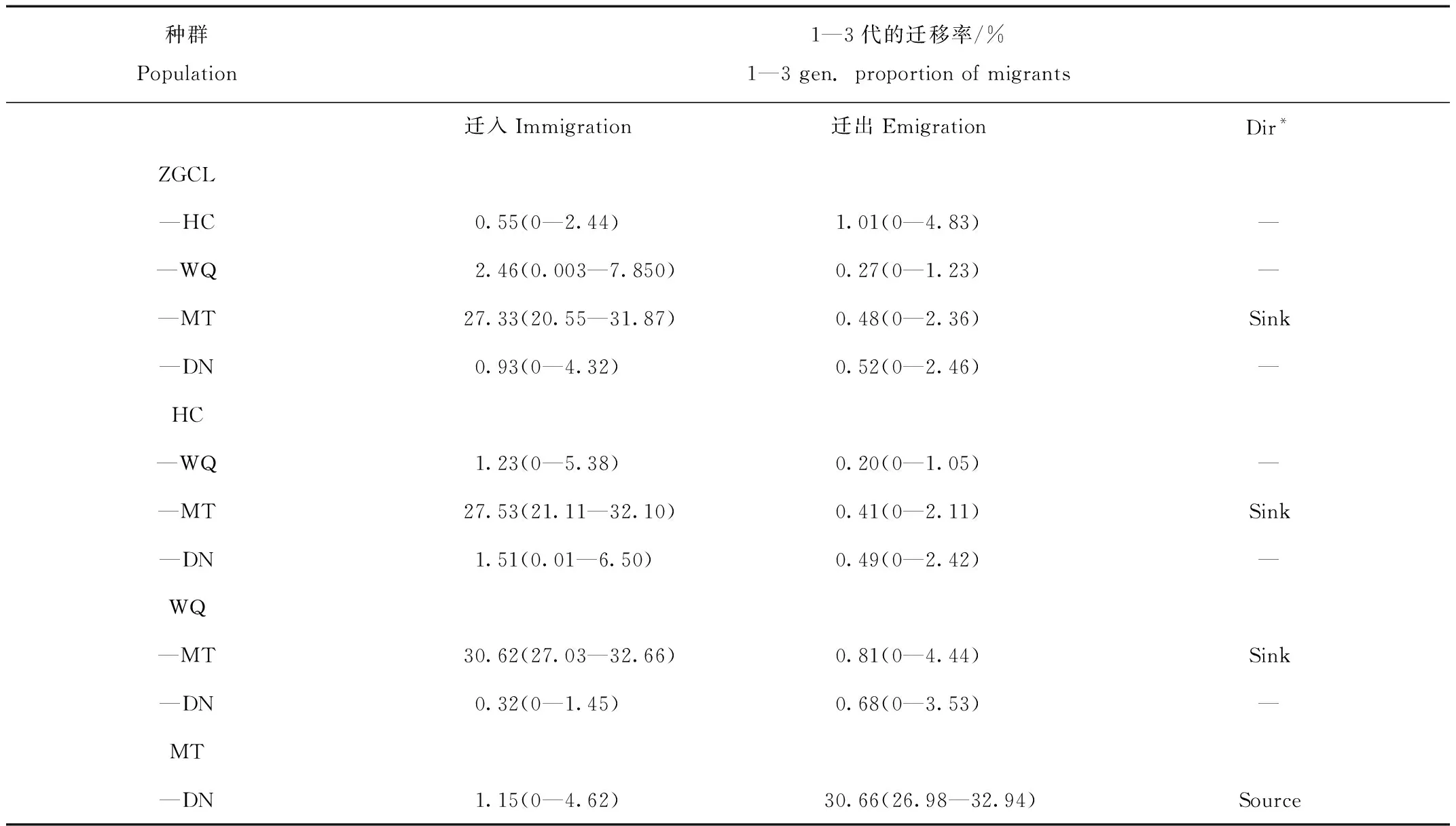
表5 种群间定向基因流评估Tab.5 Directional gene flow estimated
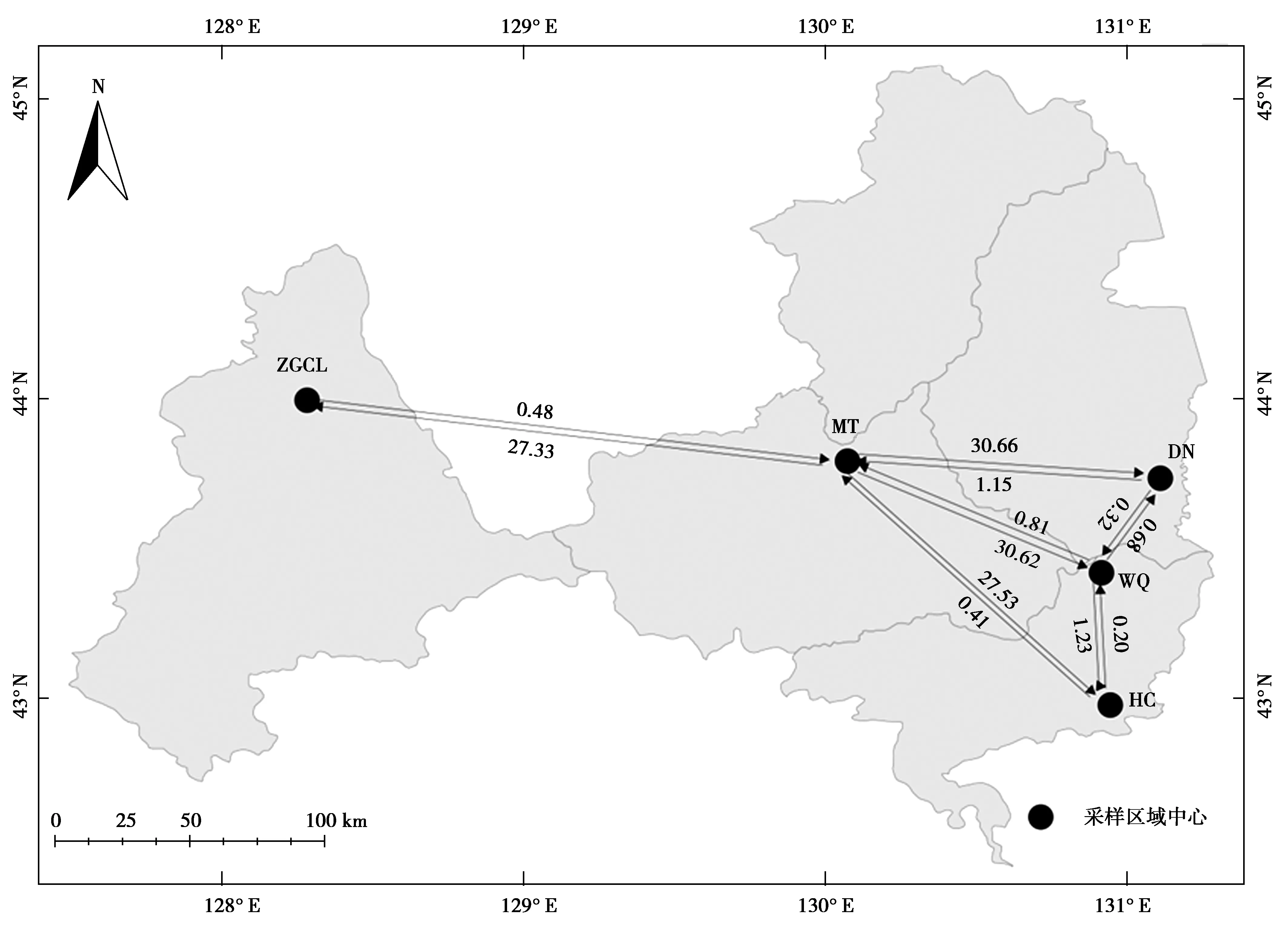
图2 基于贝叶斯方法计算的5个地理种群间的近期定向基因流Fig.2 Recent directional gene flow between 5 geographic populations calculated based on Bayesian method
3 讨论
在本研究中,调查了长白山北部西伯利亚狍局域种群的遗传多样性和种群间基因流。我们检测到高遗传多样性(He=0.714—0.732;表3),虽低于德国北部欧洲狍微卫星多样性(He=0.74—0.79)[22],但仍高于亚洲大部分西伯利亚狍种群(He=0.522—0.628)[9]。不同于盛清宇等[23]使用10个微卫星位点对长白山北部区域西伯利亚狍局域种群遗传特征的分析的研究,本研究中使用9个微卫星位点,且对地理种群的划分与其不同,虽各种群遗传多样性参数略有不同,但研究结果均表明长白山北部区域西伯利亚狍局域种群遗传多样性高。估算群体间的遗传分化系数可以区分群体间和群体内相对遗传变异大小,揭示群体遗传变异的主要来源[24]。本研究中,长白山北部区域狍种群不同地理群体间的遗传分化水平很低(FST=-0.001 1—0.018 0,表4),说明遗传变异绝大部分存在于群体内,群体间的遗传变异所占比例极小。
群体遗传学研究表明,引起群体产生遗传分化的主要原因是遗传漂变和自然选择[25]。基因流能够增加群体内遗传变异量,减少群体间的分化,使群体趋向于一致,其与遗传漂变的作用是相互撷抗的[26]。在自然群体中,群体的分化差异程度与群体间的基因流大小紧密相关,即如果不同群体间的基因流强度越高,则该群体间的分化差异程度则相对越小[27-28]。本研究中各地理种群间存在广泛的不对称的基因流,抑制了遗传漂变的作用,从而使得群体间的遗传分化非常小。虽然还需要进行更多的测试,但本研究表明长白山北部狍局域种群似乎并没有遗传差异,基因持续交流是导致种群间遗传分化低的主要原因。如果允许西伯利亚狍的连通性持续不变,我们可以预期在长白山北部将会继续扩散,甚至不断扩大。然而,需要注意的是,本研究仅探究了各种群间的基因流情况,但是何种原因导致这种种群间的交流,需要进一步分析研究。
4 结论
本研究表明长白山北部狍局域种群具有较高的遗传多样性,遗传信息丰富。其杂合度水平和等位基因丰富度高于亚洲其他地区的西伯利亚狍种群。5个局域种群之间,遗传多样性比较接近,未出现遗传多样性突高的情况。种群间存在不对称基因流,且表明较大的基因流是长白山北部狍不同地理种群间遗传分化水平很低的主要原因。本研究表明,研究区域内的西伯利亚狍种群具有良好的遗传潜力,是可以进行持续发展的种质资源。因此,本研究为西伯利亚狍种群的科学管理提供了理论依据。
致谢:感谢国家自然科学基金(31872241)、中央高校基础研究基金(2572017PZ14)和有蹄类食性分析中植物角质细胞人工智能识别研究项目(201910225555)对本研究项目的资助。感谢宁瑶博士、温都苏博士、佘雯硕士、王姣硕士给予的帮助。野外采样工作得到了项目区各林业局和保护区工作人员的支持和帮助,在这里一同表示感谢。
猜你喜欢
烟台大学学报(自然科学与工程版)(2022年3期)2022-06-30
水产科学(2022年2期)2022-03-20
南京师范大学学报(工程技术版)(2021年2期)2021-10-20
智慧健康(2021年17期)2021-07-30
红领巾·成长(2019年3期)2019-04-16
新课程·下旬(2018年9期)2018-11-14
青少年科技博览(中学版)(2015年10期)2015-01-11
中学生物学(2008年6期)2008-08-29
