
印加孔雀草对土壤细菌群落多样性的影响
2020-08-20 09:07郓玲玲张瑞海宋振付卫东王然王忠辉张国良
生态环境学报 2020年5期
郓玲玲,张瑞海,宋振,付卫东,王然,王忠辉,张国良
中国农业科学院农业环境与可持续发展研究所,北京 100081
外来入侵杂草对生物多样性、生态环境以及人类生产生活造成了严重的影响(Montserrat et al.,2011;万方浩等,2002;闫小玲等,2014)。在入侵植物与土著植物的种间竞争中,土壤微生物可能起到了重要的“桥梁”的作用,外来植物可能通过改变入侵地土壤微生物群落结构与功能促进自身生长或阻碍共生植物的生长和更新(Yu et al.,2005;Li et al.,2006)。大量研究表明,入侵植物可以通过改变土壤微生物功能类群来改变养分循环及其他环境条件,进而形成有利其竞争的微生态环境,以利于快速形成自身群落,最终实现入侵(Katharine et al.,2006;彭鑫怡等,2019;Niu et al.,2007;宋振等,2016)。入侵植物紫茎泽兰(Eupatorium adenophorum)根系可从根际土壤中选择性富集梭状芽胞杆菌(Clostridium)和肠杆菌(Enterobacter),这些根际微生物的变化使土壤营养成分更有利于紫茎泽兰生长(Chen et al.,2019);菊科植物黄顶菊(Flaveria bidentis)入侵导致土壤中链霉菌(Streptomyces)、中华杆菌(Saprospiraceae)等细菌的丰度增加,而芽孢杆菌(Bacillus)、Solibacterales等细菌的丰度减少,这些微生态环境的改变可能会影响本地植物的生长发育,从而有利于黄顶菊的入侵、竞争与扩张(Song et al.,2017);飞机草(Chromolaena odorata)可以富集入侵地土壤中的土传致病真菌半裸镰刀菌(Fusarium semitectum),使其能够抑制当地植物的生长,促进自身的入侵过程(Vaisakh et al.,2012)。另外,植物之间的竞争作用也会改变土壤细菌群落结构(Garbeva et al.,2004),如闫素丽等(2011)研究发现,黄顶菊与高丹草(Sorghum bicolor×S.sudanense)、向日葵(Helianthus annuus)、紫花苜蓿(Medicago sativa)和黑麦草(Lolium perenne)4种替代植物混合种植可以改变黄顶菊入侵地的微生物群落结构,使其接近于替代植物的土壤微生物类群,这可能是替代植物对黄顶菊具有较强竞争优势的原因所在;李光义等(2013)研究发现不同搭配方式的白三叶(Trifolium repens)、杂交甜高粱(Sorghum dochna)、杂交狼尾草(Pennisetum alopecuroides)与入侵杂草胜红蓟(Ageratum conyzoides)混种对土壤细菌多样性产生了不同程度的影响。
印加孔雀草(TagetesminutaL.)又名臭罗杰,属菊科(Asteraceae),万寿菊属(Tagetes),一年生草本植物,原产自南美洲南部,后引种到欧洲、亚洲、非洲、马达加斯加岛、印度、澳大利亚以及夏威夷等地区。在我国境内,2006年首次台湾地区发现印加孔雀草(Wang et al.,2006),2011年在北京郊区发现了印加孔雀草野生群落(张劲林等,2014),并呈现种群入侵扩张趋势,近几年陆续在河北、山东、山西、西藏等多地发生大面积危害(张瑞海等,2019)。印加孔雀草对生境的适应能力强,耐干旱、耐盐碱、耐瘠薄(Hulina,2008);具有强大的繁殖能力,种子小、产量高、且易传播(张劲林等,2014);并分泌化感物质抑制其伴生植物的生长(Meissner et al.,2013)。目前国内对其研究较少,研究内容主要集中在形态学、发生危害等方面,张瑞海等(2019)分析证明了印加孔雀草为具有危害性和侵入性的物种,土艳丽等(2019)研究表明了恶性入侵植物印加孔雀草能显著增加农作物藏青稞的死亡率;而国外对印加孔雀草的研究主要集中在其精油的化学成分及利用等方面(Martín et al.,2005)。
目前国内外对于印加孔雀草入侵对根际土壤微生物群落的影响研究尚未见报道。印加孔雀草入侵怎样影响土壤细菌群落结构?而与其共生植物存在竞争时又是否会造成土壤微生物群落结构的变化?基于以上问题,本研究采用第二代高通量测序技术,以印加孔雀草为研究对象,探讨其入侵及与本地植物竞争条件下土壤微生物群落的响应,比较分析印加孔雀草对土壤细菌群落多样性的影响及其与环境因子间的关系,以期从微生态学的角度探究印加孔雀草的入侵机制,为其综合防控及生态系统的修复提供理论依据。
1 材料与方法
1.1 试验材料采集
试验所需印加孔雀草种子、入侵发生地原位土壤皆采集于北京市昌平区兴寿镇桃林村(40°13′47.98″N、116°25′26.63″E),该地位于北京市西北部,属暖温带,为半湿润大陆性季风气候,具有明显的干季和雨季,四季分明,是印加孔雀草入侵较为严重的地区之一。所采集的原位土壤为适合印加孔雀草生存但未入侵过的褐土,万寿菊种子购买于市面种子公司。
1.2 试验设置及取样
试验区位于中国农业科学院农业环境与可持续发展研究所顺义农业环境综合试验基地(北京市顺义区 40°14′55.42″N,116°39′23.10″),设置 4 个处理(花盆大小为:23 cm×18.4 cm):未种植任何植物的裸土(不种植任何植物,记为CK)、本地植物万寿菊单优群落(只种植万寿菊,记为 Te)、入侵植物印加孔雀草与本地植物万寿菊混种群落(印加孔雀草与万寿菊1:1种植,记为TmTe)、入侵植物印加孔雀草单优群落(只种植印加孔雀草,记为Tm),每个处理3次重复。整个试验持续50 d,覆盖植物发芽、营养生长等过程,使其形成相对稳定的根际微环境,期间保证适当的水分供应。试验结束后,将每个花盆轻轻去除上层土(3 cm左右),然后采用抖土法取得植物根际土壤(Hofman et al.,1989)。采集到的每个处理的土壤样品充分混匀后,分别采用四分法取得一部分土壤样品,装进自封袋,立即带回实验室放入−80 ℃冰箱中,用于高通量测序;其他部分也分别装进自封袋,带回实验室,置于阴凉处自然晾干,用于测土壤 pH值、全氮(TN)、全磷(TP)、全钾(TK)和有机质(OM)等指标。而后随机选取各处理中(CK除外)长势一致具有代表性的5株植株(混种处理中每种植物各选取5株),将植株洗净,用于植物生物量的测定。
1.3 植物生物量及土壤理化性质的测定
将植株洗净,用滤纸吸去多余水分,120 ℃杀青、80 ℃烘干至恒质量,测定植物干质量。检测前处理后的土壤样品理化性质,包括土壤pH值(电极法),有机质(重铬酸钾容量法)、全氮(半微量开氏法)、全磷(碳酸氢钠钼蓝法)以及全钾(火焰光度法)含量,测定方法参照鲍士旦(2005)方法。
1.4 土壤DNA的提取及细菌16S rDNA的PCR扩增
采用土壤基因组 DNA提取试剂盒(北京,Solarbio公司)提取土壤样品总基因组DNA,利用1%琼脂糖凝胶电泳及超微量紫外分光光度法检测DNA纯度。以纯化后的DNA为模板,以细菌16S rDNA V3-V4区通用引物(V3F:5'-TACGGRAGGCA GCAG-3′,V4R:5′-AGGGTATCTAATCCT-3′)进行PCR 扩增(Peiffer et al.,2013;Bortolini et al.,2016)。采用TransGen AP221-02 PCR反应体系:TransStart Fastpfu DNA Polymerase,20 μL,DNA 模板 10 ng,上、下游引物(5 μmol·L−1)各 0.4 μL,dNTP(2.5 mmol·L−1) 2 μL , FastPfu Polymerase 0.4 μL ,5×FastPfu buffer 4μL,补充 ddH2O 至 20 μL。PCR扩增程序为:95 ℃预变性2 min;95 ℃变性30 s、55 ℃退火30 s,72 ℃延伸45 s,30个循环;最后于72 ℃延伸10 min,4 ℃保存。每个样品3个重复,将同一样品的PCR产物混合后用质量体积比为2%的琼脂糖凝胶进行电泳检测。
1.5 Miseq文库构建及测序
样品质检合格之后,构建测序文库,其步骤分为:(1)连接“Y”字形接头;(2)使用磁珠筛选去除接头自连片段;(3)利用PCR扩增进行文库模板的富集;(4)氢氧化钠变性,产生单链DNA片段。利用Illumina公司的Miseq PE300平台进行测序(由北京奥维森基因科技有限公司提供),步骤如下:(1)DNA片段的一端与引物碱基互补,固定在芯片上;(2)另一端随机与附近的另外一个引物互补,也被固定住,形成“桥(bridge)”;(3)PCR扩增,产生DNA簇;(4)DNA扩增子线性化成为单链;(5)加入改造过的DNA聚合酶和带有4种荧光标记的dNTP,每次循环只合成 1个碱基;(6)用激光扫描反应板表面,读取每条模板序列第一轮反应所聚合上去的核苷酸种类;(7)将“荧光基团”和“终止基团”化学切割,恢复3′端粘性,继续聚合第2个核苷酸;(8)统计每轮收集到的荧光信号结果,获知模板DNA片段的序列。
1.6 数据分析
Miseq测序得到的PE reads首先根据overlap关系进行拼接,同时对序列质量进行质控和过滤。由于不同样本间的 reads数不同,因此在分析多样性和群落组成前,先依据最低 reads数进行标准化。区分样品后进行OTU(operational taxonomic unit)聚类分析和物种分类学分析,基于 OTU进行物种多样性指数分析。基于分类学信息,在各个分类水平上进行群落结构的统计分析。在上述分析的基础上,进行一系列群落结构和系统发育等的统计学和可视化分析。使用FLASH软件(FLASH v 1.2.7)对下机数据进行拼接,得到OTUs数据(Raw Tags),利用Trimmomatic(v 0.33)对Raw Tags质控过滤,得到Clean Tags,所得Clean Tags数据上传至在美吉云平台 I-Sanger(https://www.i-sanger.com/),进行数据分析。基于 97%相似度下,利用 Usearch(vsesion 7.0)软件在Silva数据库进行OTU物种分类学分析和聚类分析。4组样本利用Adonis置换多因素方差分析和 ANOSIM 相似性分析,判断分组的可信度和可行性。
Ace、Chao指数为土壤细菌群落丰富度指标,皆用以估计群落中OTU数目,Chao指数采用chao1算法(Chao,1984),Ace指数算法与其不同(Chao et al.,1993);Shannon、Simpson 指数为用来估算样品中细菌群落多样性指标,Shannon值越大群落多样性越高(Shannon,1948a,1948b),Simpson指数则是由 Edward Hugh Simpson(1949)提出,Simpson值越大群落多样性越低。利用 mothur(version v.1.30.1)对 Ace、Chao、Shannon 和 Simpson指数进行分析Alpha多样性,用T检验分析Alpha多样性指数差异,然后用R作图;利用柱形图表示细菌群落组成;各样本差异物种组成通过One-way ANOVA单因素方差分析,LEfSe软件首先使用nonparametric factorial Kruskal-Wallis (KW) sum-rank test(非参数因子克鲁斯卡尔—沃利斯秩和验检)检测具有显著丰度差异特征,并找到与丰度有显著性差异的类群,然后采用线性判别分析(LDA)来估算每个组分(物种)丰度对差异效果影响的大小;环境因子与菌群的关联分析,检测pH、OM、TN、TP、TK等5个指标,利用单因素方差分析及posthoc检验筛选不同样点根际土壤菌群差异物种,通过计算环境因子与根际土壤菌群之间的相关性系数(Spearman等级相关系数、Pearson相关系数等),然后利用Pheatmap Package软件,将获得的数值矩阵通过Heatmap图展示出平均相对丰度前10的属与环境因子的相关性。
2 结果
2.1 植物生物量及土壤理化性质
各处理印加孔雀草与万寿菊的生物量及土壤理化性质指标分析结果如表1所示。结果显示,无论单种(Tm、Te)处理还是混种(TmTe)处理,入侵植物印加孔雀草的生物量都显著高于万寿菊。土壤理化性质结果显示,相比于CK,3个处理(Tm、Te、TmTe)的土壤理化性质都发生了不同程度的变化。土壤pH均呈碱性,且有印加孔雀草入侵过的土壤pH显著增加,各处理之间差异显著(P<0.05);种植植物的3个处理中,TmTe的土壤全氮(TN)质量分数高于Tm(P=0.033)与Te(P=0.722),均值为0.116%;Tm的土壤全钾(TK)质量分数显著高于 TmTe(P=0.047)与 Te(P=0.028),为 1.473%;土壤全磷(TP)、有机质质量分数在各处理之间差异不显著。
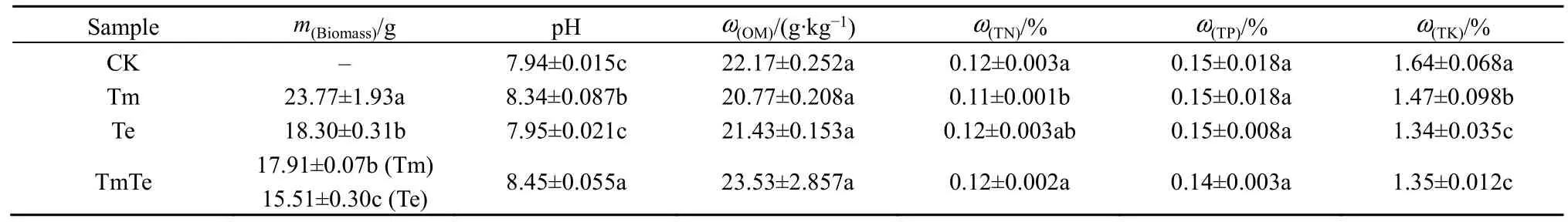
表1 植物生物量及土壤理化性质比较Table 1 Comparison of plant biomass, soil physical and chemical properties
2.2 土壤细菌的OTU丰度和α多样性
对 12个样品进行高通量测序,共产生 223.72 Mb干净数据,经过拼接和过滤处理后,获得 16S rDNA标签序列,并根据97%的序列相似性划为不同的OTU。OTUs丰度稀释曲线(图1)显示,随着测序数量的增加,稀释曲线斜率逐渐降低,趋向平坦但未达平台期,说明大多数样本的测序数据量足够,能够反映样品中的物种组成特征。
图1 样品在遗传距离0.03下的稀释曲线图Fig. 1 The rarefaction of different samples obtained at the genetic distances of 0.03
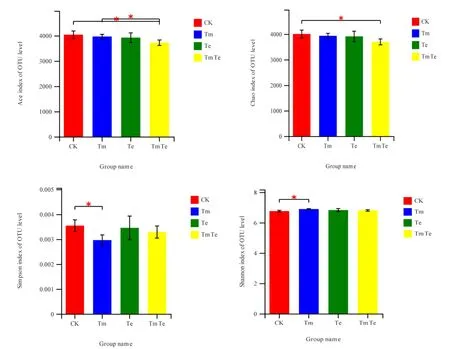
图2 样品在遗传距离0.03下的细菌α多样性指数Fig. 2 Alpha diversity for different samples obtained at genetic distance of 0.03
土壤细菌丰富度指数和多样性指数如图 2所示,CK、Tm、Te与TmTe的Simpson指数均值分别为0.0035、0.0030、0.0035、0.0033,Shannon指数均值分别为6.75、6.89、6.82、6.80,Tm与CK表现出了明显差异,Simpson指数显著低于 CK(P=0.033)、Shannon指数显著高于CK(P=0.024),说明印加孔雀草单优群落的细菌群落多样性最高,其它处理未表现出与CK的显著性差异;CK、Tm、Te以及 TmTe的 Ace指数均值分别为 4035.9、3964.7、3918、3718.2,Chao指数的均值分别为4000.4、3928.6、3907.2、3691.7,TmTe的Ace指数显著低于CK(P=0.039)与Tm(P=0.035)、Chao指数显著低于CK(P=0.049),说明印加孔雀草与万寿菊混种处理群落丰富度最低,印加孔雀草单优群落的丰富度高于混种处理,万寿菊单优群落与其他处理未表现出显著性差异。
2.3 高通量序列生物信息学分析
通过测序共产生4615个细菌OTU(图3),其中 4个处理共有的 OTUs为 3169个,占总数的68.67%。CK的细菌群落数最高,平均含4189个细菌OTU;Tm次之,为4110个OTU;TmTe的细菌群落数最低,为3814个OTU。且TmTe和Te相比,TmTe特有的细菌OTU数目为321个,占8.42%。结果说明,不同处理带来了不同程度的土壤细菌OTU差异,混种处理土壤细菌OTU数目最低,印加孔雀草单优群落处理有使OTU数目升高的趋势,这与α多样性中的群落丰富度的研究结果一致。
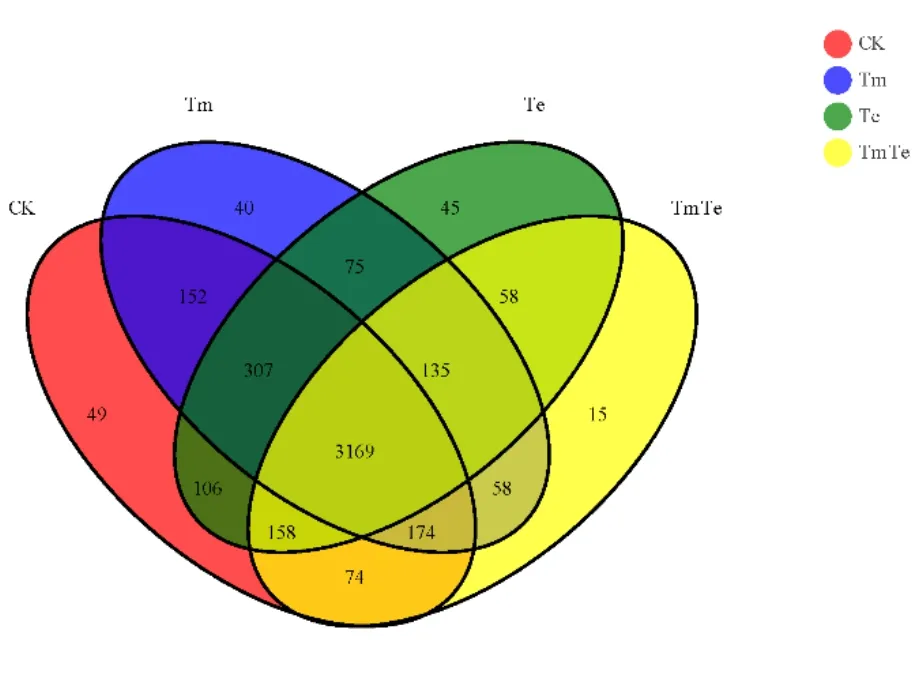
图3 细菌OTUs分布韦恩图Fig. 3 Venn graph of bacteria OTUs distribution
2.4 细菌群落组成
选取各样品中细菌在门分类水平上最大丰度排名前 10的物种,生成物种相对丰度堆积图(图4)。由图4可知,4个处理的土壤样品都具有丰富的物种,其中丰度最高的 5个门为变形菌门(Proteobacteria)、酸杆菌门(Acidobacteria)、放线菌门(Actinobacteria)、绿弯菌门(Chloroflexi)及芽单胞菌门(Gemmatimonadetes),共计占各处理的80%以上。12个样品中所检测到的细菌门的数量相似,但不同处理不同门所占的比例不同。相比于裸土(CK),其他3个处理中不同门所占比例都有不同程度的变化。进一步分析3个处理(Te、TmTe、Tm)的群落组成平均值,发现随着印加孔雀草密度的增加(Te、TmTe、Tm),放线菌门、绿弯菌门以及芽单胞菌门所占的比例呈现出先升高再降低的趋势,变形菌门呈现先降低后升高的趋势,而酸杆菌门呈现不断上升的趋势,但差异不显著。这表明不同门下的细菌在印加孔雀草入侵过程中发挥着不同的功能,而酸杆菌门有可能在其入侵过程中起到比较重要的作用。
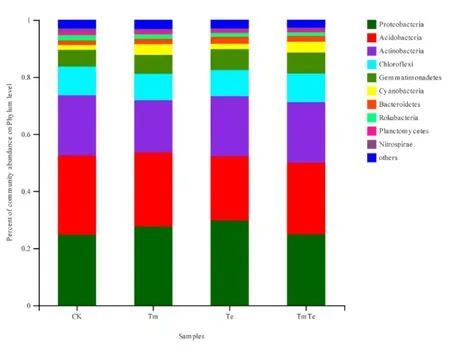
图4 门水平上的细菌相对丰度柱形图Fig. 4 The relative abundance of bacteria in Phylum level
2.5 Lefse多级物种差异判别分析
抽取出在门至属水平上各处理序列数总和大于等于100个OTU的细菌,利用LEfSe软件,可以得到Tm、Te、TmTe分别与CK相比显著富集且具有显著差异的菌,这些细菌与各自处理的植物群落的生长与维持关系密切,分别记为Tm vs CK、Te vs CK和 TmTe vs CK,对其进行韦恩图分析(图5)。得到对印加孔雀草入侵有利的细菌,为共同存在于Tm vs CK和TmTe vs CK中但不属于Te vs CK的10个OTU类群。对其进行进一步分析(表2)显示,这10个OTU类群隶属于变形菌门、放线菌门、蓝细菌门(Cyanobacteria)及酸杆菌门4个门,其数量分别为3、3、3、1个。其中,变形菌门下的有多囊粘细菌科(Polyangiaceae)、侏囊菌科(Nannocystaceae)等3个科下的3个菌属;放线菌门下的有假诺卡氏菌科(Pseudonocardiaceae)、Ilumatobacteraceae科等2个科下的3个菌属;蓝细菌门下的有念珠藻科(Nostocaceae)、席藻科(Phormidiaceae)等2个科下的 3个菌属以及酸杆菌门下Blastocatellaceae科下的 1个菌属。另外,TmTe显著性差异于其他处理的菌为图中 29个OTU类群,Tm显著性差异于其它处理的菌为图中33个OTU类群。
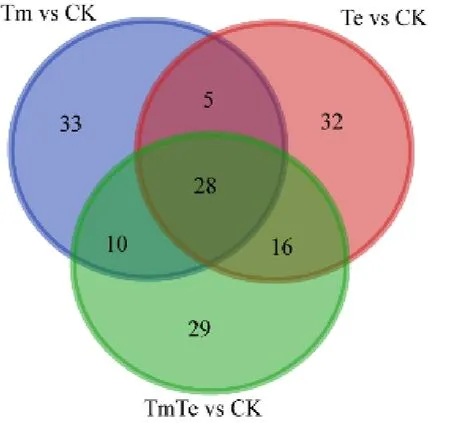
图5 3个处理分别显著富集且具有显著差异于CK的细菌韦恩图Fig. 5 Venn graph of bacteria with significant enrichment and difference between the three treatments and CK
2.6 环境因子关联分析
选取各样品中细菌在门分类水平上最大丰度排名前 10的物种,通过计算环境因子与菌群之间的 Spearman等级相关系数,生成环境因子相关性Heatmap图(图6)。由图6可知,不同菌群与各种环境因子体现出不同程度的相关性关系。其中,硝化螺旋菌门(Nitrospirae)、酸杆菌门、棒状杆菌门(Rokubacteria)、浮霉菌门(Planctomycetes)、放线菌门、变形菌门、拟杆菌门(Bacteroidetes)、绿弯菌门以及芽单胞菌门与土壤全磷、全钾含量呈不同程度的正相关,与土壤pH呈现出了不同程度的负相关,与土壤全氮和土壤有机质含量无明显相关性,个别呈现出了微弱的负相关关系。其中,放线菌门与土壤全钾含量呈极显著正相关(R=0.71735,P=0.00863),硝化螺旋菌门、变形菌门、拟杆菌门、绿弯菌门以及芽单胞菌门与土壤全钾含量呈显著正相关。但是,蓝细菌门(Cyanobacteria)与环境因子之间的关系不同于其他菌门,具体表现与土壤全氮、全磷、全钾含量呈微弱的负相关,与pH、有机质含量呈微弱的正相关。
3 结论与讨论
3.1 讨论
高通量序列生物信息学及α多样性分析结果表明,印加孔雀草单优群落的土壤细菌群落多样性最高且显著高于CK,其它处理未表现出与CK的显著性差异,这说明印加孔雀草的入侵可以提高土壤细菌群落多样性,且入侵植物印加孔雀草对于土壤细菌多样性的作用效果要大于与其共生的万寿菊。宋振等(2016)在研究同为菊科的入侵植物黄顶菊的根际微生物群落结构时发现,黄顶菊单优群落的土壤细菌群落多样性显著高于本地植物,这与本研究的结果是一致的;入侵植物空心莲子草(Altemanthera philoxeroides)可以通过改变土壤微生物菌群的结构和丰富度从而影响土壤有机碳的形成,其入侵提高了土壤微生物群落的多样性(李珂,2019),印加孔雀草提高土壤细菌群落多样性从而形成有利于自身生长的微生态环境,进而提高生物量以促进自身生长及发育,这很有可能是其入侵成功的重要原因之一。有研究表明,紫茎泽兰与本地植物存在竞争时,其土壤细菌的群落丰富度与未入侵土壤及紫茎泽兰单优群落土壤之间未表现出明显差异(朱珣之等,2015);互花米草(Spartina alterniflora)入侵改变了土壤微生物群落和提高了根际微生物丰富度,而高丰富度的根际微生物影响了土壤养分循环和植物间的竞争关系,从而促进互花米草快速生长,并成功入侵(章振亚等,2012)。在本研究中,印加孔雀草与万寿菊混种处理的群落丰富度最低且显著低于 CK,印加孔雀草、万寿菊单种处理都未表现出与CK的显著性差异,说明印加孔雀草与万寿菊种间竞争使得土壤细菌群落丰富度降低,这可能是万寿菊与印加孔雀草相互作用的结果。

表2 在印加孔雀草入侵过程中起重要作用的10种菌在各水平上的分类及数目Table 2 Classification and the number in 10 bacteria that play an important role in invasion process of T. minuta L.
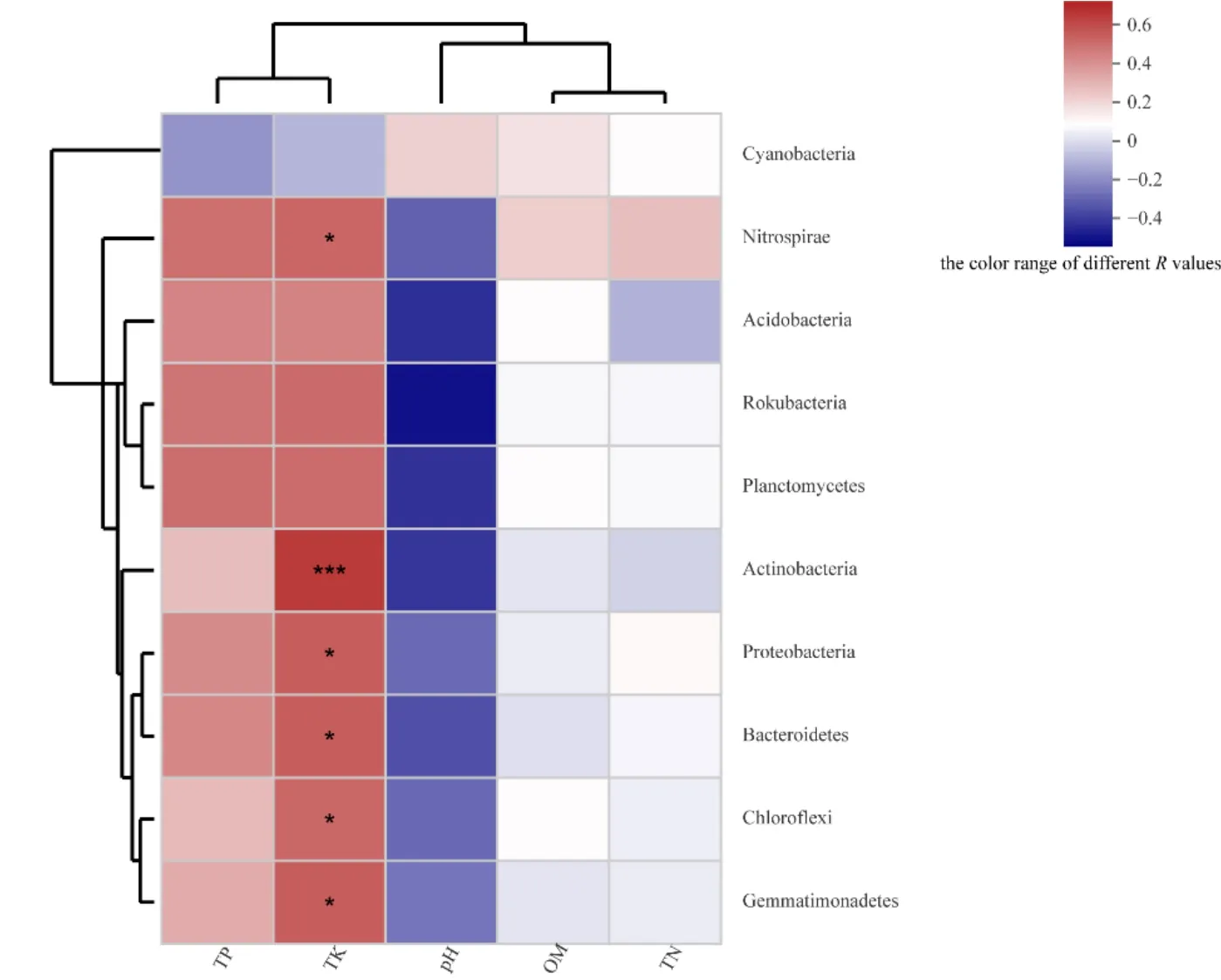
图6 门分类水平上最大丰度排名前10的细菌与环境因子的相关性Heatmap图Fig. 6 Heatmap map of the correlation between bacteria and environmental factors in the top 10 most abundant species on Phylu m level
本研究各处理中丰度最高的5个门依次为变形菌门、酸杆菌门、放线菌门、绿弯菌门和芽单胞菌门。通过 Lefse多级物种差异判别以及进一步的Venn分析,得到共同存在于Tm vs CK和TmTe vs CK中但不属于Te vs CK的类群,为4个门(变形菌门、蓝细菌门等)下的8个科(多囊粘细菌科、侏囊菌科、念珠藻科等)的共10个OTU类群,这些类群可能在印加孔雀草在入侵过程中起着重要作用。例如,多囊粘细菌科与侏囊菌科属于粘细菌,隶属于变形菌纲的粘球菌目。粘细菌是一类捕食菌,也是继放线菌和真菌之后又一重要的活性次级代谢产物产生菌(王春玲等,2019),广泛分布于全球各地的土壤中,能够形成对温度、干旱、辐射、盐胁迫均具有抗性的粘孢子,而具有较高的环境适应力(Shimkets,1991),其群落和代谢产物对土壤微生态环境的改变很可能有利于印加孔雀草的入侵。而与TmTe显著差异的存在于Tm、Te的29个OTU类群可能有利于维持印加孔雀草入侵的土壤微生态系统结构,Tm显著性差异于 Te、TmTe的33个OTU类群则可能有利于印加孔雀草单优群落状态的维持,但是否是这些 OTU类群及其功能还需要进一步深入研究。
不同门类的细菌同时受到土壤理化性质和植被特性等环境因子的影响而产生差异(Zhou et al.,2017),本研究中环境因子关联分析结果表明,菌群与环境因子存在不同程度的显著相关性,印加孔雀草入侵后,其根际土壤可能会快速形成稳定的细菌群落结构以及多样性,有利于提高印加孔雀草其自身对环境的适应能力以及单优群落的形成。环境因子关联分析也表明,不同门下的菌群与各种环境因子体现出不同的相关性特征,放线菌门与土壤全钾含量呈极显著正相关,硝化螺旋菌门、变形菌门、拟杆菌门、绿弯菌门以及芽单胞菌门与土壤全钾含量呈显著正相关,而蓝细菌门与环境因子的关系则表现出了和其他菌门较为不同的特点。蓝细菌是发现的最早能够利用光合作用释放氧气的原核生物(Bryant,1995),在自然界中不仅分布在淡水、海水等水体中,也广泛分布在各类土壤以及极端环境中(Nicholas et al.,1992)。本研究中蓝细菌表现出与环境因子特殊的相关性可能是由于印加孔雀草在快速繁殖扩散过程中对土壤有机物合成和氧气释放有着较为特殊的需求。
植物根系分泌的很多次生代谢物质可作为植物与土壤微生物互作的信号分子物质而影响植物根际土壤微生物群落结构(Ueda et al.,2010;Ahuja et al.,2012),如豚草的次生代谢产物对土壤微生物具有一定的选择抑制作用(赵昕,2003),黄顶菊次生代谢产物紫云英苷可以改变土壤微生物群落结构(张瑞海,2017)等。目前已有研究发现,印加孔雀草植株能够分泌萜烯类化感物质从而抑制伴生植物的生长(Liza et al.,2008;Scrivanti et al.,2003),是否这些物质也导致了印加孔雀草土壤微生物的改变从而有利于自身群落的稳定,或者有其他特殊的次生代谢产物影响了土壤微生物群落,需要进一步深入研究。另外,本研究采用了入侵地未生长过印加孔雀草的单一褐土类型土壤,然而不同土壤类型的起始土壤细菌群落的组成存在差异,这种差异很可能会影响入侵种对土壤的响应,印加孔雀草对不同土壤类型的响应是否与本试验结果存在不同,这还需要大量重复试验进行研究。
3.2 结论
印加孔雀草入侵能够改变入侵地土壤细菌多样性。印加孔雀草显著提高了土壤细菌群落多样性,其与万寿菊混种使得土壤细菌群落丰富度显著降低,且印加孔雀草竞争效果大于非入侵植物万寿菊;土壤中细菌相对丰度最高的前5个门分别是变形菌门、酸杆菌门、放线菌门、绿弯菌门和芽单胞菌门;不同菌群与环境因子表现出了不同程度的正负相关关系,其中放线菌门、硝化螺旋菌门、变形菌门、拟杆菌门、绿弯菌门以及芽单胞菌门与土壤全钾含量呈显著正相关,蓝细菌门和环境因子的相关关系不同于其他菌门;多囊粘细菌科、侏囊菌科、念珠藻科等8个科的10个OTU类群有可能在印加孔雀草入侵过程中起着重要作用。本研究结果为揭示印加孔雀草入侵的土壤微生态机制提供了理论依据。
猜你喜欢
中国水土保持(2022年2期)2022-04-07
云南农业(2021年9期)2021-09-24
文萃报·周五版(2020年37期)2020-10-12
中国比较医学杂志(2020年4期)2020-05-26
意林·作文素材(2019年21期)2019-12-19
参花(下)(2019年8期)2019-09-11
看天下(2019年14期)2019-06-11
中国沼气(2019年1期)2019-04-13
生物安全学报(2019年3期)2019-02-15
吉林农业(2018年23期)2018-01-17
