
HPLC 测定左乙拉西坦氯化钠注射液中异构体的含量
2020-11-18 06:27段云
质量安全与检验检测 2020年5期
段云
(乌兰察布医学高等专科学校 内蒙古乌兰察布 012000)
1 前言
左乙拉西坦是当前报道的唯一具有预防癫痫发病的抗癫痫药品,于1999 年首次被美国食品药品监督管理局(FDA)批准在美国上市。由于效果好、副作用低,其在全球范围内的应用越来越广,目前主要用于年龄大于或等于1 个月的儿童与成人癫痫患者部分性发作的辅助治疗药[1-3]。 左乙拉西坦氯化钠注射液是由左乙拉西坦(C8H14N2O2)和氯化钠组成的注射液制剂,对于暂时不能口服左乙拉西坦的癫痫患者,本品提供了一个很好的选择。 左乙拉西坦为手性药物,其右旋异构体-右乙拉西坦(图1)对于抑制癫痫发作只有轻微或不明显的药效作用,因此需控制其右旋异构体的含量。 目前有关左乙拉西坦中异构体的测定已有报道,但对其制剂中异构体的检测文献很少[4-6]。 本文建立了左乙拉西坦氯化钠注射液中异构体含量的高效液相色谱法,该方法简便、灵敏、准确、重现性好,可为左乙拉西坦氯化钠注射液异构体的准确定量提供参考。
2 仪器与试剂
2.1 仪器
电子天平(梅特勒AB135-S);高效液相色谱仪(日本岛津-紫外检测器高效液相色谱仪,色谱柱为CHIRALPAK IC 柱,250 mm×4.6 mm,5 μm)。

图1 右乙拉西坦的分子式
2.2 试剂
无水乙醇(色谱纯,MREDA);乙酸铵(分析纯);左乙拉西坦氯化钠注射液 (小试批样品批号:20130501、20130504、20130507; 中试批样品批号:20131201、20131202、20131203;北大医院重庆大新药业有限公司)。
3 方法与结果
3.1 色谱条件
采用 CHIRALPAK IE 色谱柱(250 mm×4.6 mm,5 μm),以 10 mmol 乙酸铵水溶液(乙酸铵 0.77 g,加水 1 000 mL 溶解)-无水乙醇(80∶20)为流动相,流速0.4 mL/min,检测波长为215 nm,柱温35℃,进样体积 10 μL,运行 30 min,进行测定。
3.2 线性关系考察
贮备液:精密称取左乙拉西坦对照品10.03 mg,置于100 mL 量瓶中,加流动相溶解并稀释至刻度,摇匀,作为贮备液。
左乙拉西坦线性溶液配制:分别精密吸取贮备液适量,加溶剂稀释成浓度为每1 mL 中含左乙拉西坦 0.5、1.0、2.5、5.0、7.5、10.0 μg 的溶液。
精密吸取上述配制溶液各10 μL 分别注入液相色谱仪,按照上述色谱条件进行测定。 以浓度(C)为横坐标(X 轴),以峰面积(A)为纵坐标(Y 轴),得到浓度和峰面积的回归方程为:Y=18.3201X+0.2909,线性相关系数(r)=0.999 9。结果表明,左乙拉西坦异构体在 0.50~10.02 μg/mL 具有良好的线性关系, 试验结果详见图2。

图2 左乙拉西坦异构体线性关系图
3.3 专属性考察
3.3.1 空白辅料干扰
称取空白辅料500 mg 规格2 mL 置于10 mL 量瓶中,加流动相稀释至刻度;空白辅料1 000 mg 规格1 mL 置于10 mL 量瓶中,加流动相稀释至刻度;空白辅料1 500 mg 规格3 mL 置于50 mL 量瓶中,加流动相稀释至刻度,作为空白辅料溶液。分别精密吸取10 μL 溶液注入液相色谱仪,进行测定。结果表明,空白辅料不干扰本品异构体检测。
3.3.2 系统适用性试验
称取左乙拉西坦消旋体适量,加入流动相制成1 mg/mL 的系统适用性溶液。 精密吸取系统适用性溶液10 μL 注入液相色谱仪进行测定。结果表明,在该色谱条件下,色谱柱的理论板数(n)较高,分离度大于1.5,符合常规测定要求,试验结果详见表1。

表1 异构体方法-系统适用性试验数据
3.4 稳定性试验
取本品适量(500 mg 规格样品 2 mL,1 000 mg规格 1 mL,1 500 mg 规格 0.6 mL),置于 10 mL 量瓶中,加流动相稀释至刻度,摇匀,作为供试品溶液。 分别于 0、2、4、6、8、10、12 h 进样检测,以检测主峰面积和异构体峰面积的变化情况衡量供试品溶液稳定性。结果表明,本品在室温放置12 h 内,主峰面积及异构体峰面积均无明显变化,说明本方法溶液稳定性良好,符合常规分析要求,试验结果详见表2。

表2 3 批样品溶液稳定性考察结果
3.5 重复性试验
取本品适量(500 mg 规格样品 2 mL,1 000 mg规格 1 mL,1 500 mg 规格 0.6 mL),置于 10 mL 量瓶中,加流动相稀释至刻度,摇匀,作为供试品溶液。 分别配制6 份,以样品中检测到的异构体峰面积为指标,验证本方法的重复性,结果表明,平行配制6 份样品,异构体杂质含量无明显变化,表明该方法重复性良好,试验结果详见表3。

表3 3 批样品溶液重复性考察结果
3.6 精密度试验
取本品适量(500 mg 规格样品 2 mL,1 000 mg 规格 1 mL,1 500 mg 规格 0.6 mL),置于 10 mL 量瓶中,加流动相稀释至刻度,摇匀,作为供试品溶液。 连续进样6 针,计算峰面积的RSD。 在该色谱条件下,左乙拉西坦注射液异构体对照溶液连续进样6 次,其保留时间及峰面积的RSD 小于2.0%,表明本色谱条件进样精密度良好,试验结果详见表4。
3.7 检测限试验
称取左乙拉西坦对照品适量制成0.1 mg/mL 溶液再加溶剂逐级稀释至适宜浓度,在上述色谱条件下,以3 倍信噪比作为检测限,结果拟定左乙拉西坦检查浓度为1.0 mg/mL,检测限为0.3 ng,即异构体在0.003%以上可被检出,表明本品色谱条件灵敏度高,适合异构体杂质的控制。
3.8 定量限试验
称取左乙拉西坦对照品适量制成0.1 mg/mL 溶液再加溶剂逐级稀释至适宜浓度,在上述色谱条件下,以10 倍信噪比作为定量限,结果拟定左乙拉西坦检查浓度为1.0 mg/mL,定量限为1.0 ng,即异构体在0.010%以上可定量检测,表明本品色谱条件能准确定量,灵敏度高。

表4 3 批样品进样精密度试验结果
3.9 耐用性试验
按照中国药典2015 版二部附录药品质量标准分析方法验证指导原则,进行耐用性试验。针对本品色谱条件,主要从流动相比例、流速和柱温进行耐用性试验。 考察异构体检测条件微小变化时异构体杂质的检出情况,以更直观地考察系统的耐用性。结果发现,调整色谱系统(流动相比例、流速、柱温),本品异构体杂质含量均无明显变化, 异构体和主峰的分离度大于1.5,表明本方法耐用性良好。
3.10 3 批中试样品异构体检查
取中试 3 批样品 0.5 g/100 mL 规格(20131201、20131202、20131203)、1.0 g/100 mL 规格(20131201、20131202、20131203)、1.5 g/100 mL 规格(20131201、20131202、20131203),按照上述确定的方法依次对3 个规格3 批样品的异构体进行检查,结果显示中试3 批样品中异构体杂质量均低于0.8%,符合规定,测定结果详见表5。
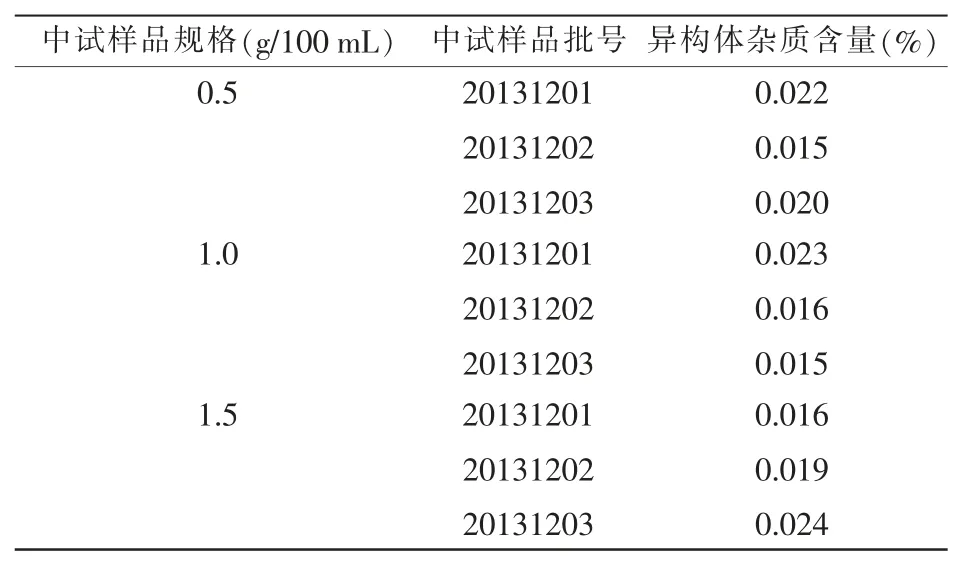
表5 异构体杂质含量测定结果
4 讨论
左乙拉西坦注射液中异构体的检查主要依据左乙拉西坦片进口标准和美国药典(USP)左乙拉西坦口服溶液,同时结合中国药典2010 年版二部附录并结合本品的性质而定。该色谱条件系统适用性好,精密度高,专属性好,试验结果显示本品异构体测定溶液稳定性好,比例对异构体杂质测定结果无影响,说明所选色谱系统可准确测定样品中的异构体。
本品中试3 批异构体杂质检测结果与原料中异构体杂质含量相比无明显变化,说明本品制剂生产过程中异构体杂质含量不发生变化。 由本品影响因素试验及加速长期稳定性结果可知,放置过程中异构体杂质含量无明显变化,考虑到本品原料中需对异构体杂质进行严格控制,故建议本品异构体杂质检查不用单独设定标准。
5 结论
用高效液相色谱法测定左乙拉西坦氯化钠注射液中异构体的含量,灵敏度高,重现性好,耐用性好,测定结果准确可靠,该法可用于测定左乙拉西坦氯化钠注射液中异构体的含量,可有效、准确控制左乙拉西坦氯化钠注射液的质量。
猜你喜欢
中国典型病例大全(2022年11期)2022-05-13
浙江化工(2022年4期)2022-05-07
健康体检与管理(2022年2期)2022-04-15
昆明医科大学学报(2021年10期)2021-12-02
临床与实验病理学杂志(2021年7期)2021-09-06
中华养生保健(2020年3期)2020-11-16
中华养生保健(2020年1期)2020-11-16
物理学报(2019年12期)2019-06-29
作文周刊·小学四年级版(2018年40期)2018-04-09
西部学刊(2017年8期)2017-09-08
