
Ipragliflozin-induced improvement of liver steatosis in obese mice may involve sirtuin signaling
2021-01-14 00:34TakayoshiSugaKenSatoTatsuyaOhyamaShoMatsuiTakeshiKobayashiHirokiTojimaNorioHoriguchiYuichiYamazakiSatoruKakizakiAyakaNishikidoTakashiOkamuraMasanobuYamadaTadahiroKitamuraToshioUraoka
World Journal of Hepatology 2020年7期
Takayoshi Suga, Ken Sato, Tatsuya Ohyama, Sho Matsui, Takeshi Kobayashi, Hiroki Tojima, Norio Horiguchi,Yuichi Yamazaki, Satoru Kakizaki, Ayaka Nishikido, Takashi Okamura, Masanobu Yamada, Tadahiro Kitamura, Toshio Uraoka
Takayoshi Suga, Ken Sato, Tatsuya Ohyama, Takeshi Kobayashi, Hiroki Tojima, Norio Horiguchi, Yuichi Yamazaki, Satoru Kakizaki, Toshio Uraoka, Department of Gastroenterology and Hepatology, Gunma University Graduate School of Medicine, Maebashi 371-8511, Gunma, Japan
Takayoshi Suga, Sho Matsui, Tadahiro Kitamura, Metabolic Signal Research Center, Institute for Molecular and Cellular Regulation, Gunma University, Maebashi 371-8512, Gunma, Japan
Ayaka Nishikido, Takashi Okamura, Masanobu Yamada, Department of Medicine and Molecular Science, Gunma Graduate School of Medicine, Maebashi 371-8511, Gunma, Japan
Abstract
Key words: Selective sodium glucose cotransporter 2; Nonalcoholic fatty liver disease; Sirtuin 1; Peroxisome proliferator-activated receptor γ coactivator 1α; Peroxisome proliferator-activated receptor α; Fibroblast growth factor-21
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD), a hepatic manifestation of metabolic syndrome, is a common chronic liver disease.It includes isolated fatty liver and nonalcoholic steatohepatitis (NASH), the latter of which can progress to cirrhosis and liver cancer in some individuals[1].This disease is associated with obesity, insulin resistance, and type 2 diabetes mellitus (T2DM).As lifestyles have become increasingly sedentary and dietary patterns have changed, the worldwide prevalence of NAFLD has dramatically increased[2].The most challenging problem is that no pharmacological therapies have been established for NAFLD so far[3].
Sodium glucose cotransporter 2 (SGLT2) inhibitors are newly developed oral antidiabetic drugs.SGLT2 is primarily expressed in the kidneys and reabsorbs approximately 90% of the glucose filtered by the renal glomeruli.SGLT2 inhibitors, which lower glucose levels independently of insulin action by facilitating the excretion of glucose in urine, are expected to become candidate therapeutic agents not only for T2DM but also for NASH/NAFLD[4,5].Ipragliflozin is a selective SGLT2 inhibitor that is orally administered.Previous reports have shown that ipragliflozin improves liver steatosis in animal models[6-8]and clinical settings[9,10].However, the mechanisms by which SGLT2 inhibitors improve liver steatosis are not fully understood.
Recently, chronic administration of an SGLT2 inhibitor was reported to drive a fuel shift, decreasing tissue glucose disposal and increasing lipid use[11].Therefore, we hypothesized that sirtuin 1 (SIRT1), a NAD+-dependent protein deacetylase with numerous substrates, might be associated with the amelioration of liver steatosis by SGLT2 inhibitors.SIRT1 plays important roles in controlling energy homeostasis and longevity in mammals[12,13].For example, SIRT1 improves sensitivity to both leptin and insulin, which act on proopiomelanocortin neurons to increase sympathetic activity toward adipose tissues and to promote the browning of white fat, and is involved in energy and glucose homeostasis[14].Pharmacological activation of SIRT1 signaling reportedly ameliorates fatty liver[15,16].In contrast, hepatocyte-specific deletion of SIRT1 impairs peroxisome proliferator-activated receptor α (PPARα) signaling, decreases fatty acid β-oxidation, and results in liver steatosis and inflammation[17].Peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α), a key coactivator for PPARα signaling[18], is known to be a direct substrate of SIRT1[19].PGC-1α interacts with multiple transcription factors to enhance mitochondrial metabolic capacity[20].Moreover, hepatic SIRT1 attenuates liver steatosis and controls energy balance by inducing the activation of fibroblast growth factor-21 (FGF21)[21].Hepatic FGF21 is regulated by PPARα and is a key mediator of hepatic metabolism[22].All of the above findings suggest that the SIRT1-PGC-1α/PPARα-FGF21 pathway is important in lipid homeostasis in the liver.
It has not been fully elucidated whether the amelioration of liver steatosis mediated by the SGLT2 inhibitor ipragliflozin is associated with SIRT1 signaling.The objectives of our study were thus to evaluate thein vivoeffects of the selective SGLT2 inhibitor ipragliflozin on liver steatosis and to investigate the mechanisms by which this SGLT2 inhibitor improves liver steatosis in obese (ob/ob) mice.In particular, the primary experimental aim was to clarify the role of SIRT1 signaling in ipragliflozin-mediated attenuation of liver steatosis inob/obmice.
MATERIALS AND METHODS
Animals and animal treatment protocol
We purchased 6-wk-old maleob/obmice and their lean sex-matched littermates from Charles River Co., Ltd.(Yokohama, Japan).All mice were kept under a 12:12 h lightdark cycle with free access to food and water.After the mice had acclimated to the rearing environment for 2 wk, they were fed a normal chow diet (CLEA Rodent Diet CE-2) from CLEA Japan, Inc.(Tokyo, Japan).The diet was changed to a normal chow diet (D12450B) from Research Diets (Tokyo, Japan) or an ipragliflozin-supplemented D12450B chow diet when the mice were 8 wk old.The treatment groups were composed ofob/obmice that were fed a normal chow diet only or a normal chow diet supplemented with one of two different doses of ipragliflozin (3 mg/kg or 10 mg/kg, Astellas Pharma Inc., Tokyo, Japan), and the control group was composed of lean littermates fed a normal chow diet.Theob/obmice were randomly assigned to the 3 treatment groups, each of which comprised 8 mice.After 4 weeks of feeding, all mice were sacrificed, total liver resection was performed, and the specimens were analyzed.For verification of SGLT2 mRNA expression, liver and kidney specimens were obtained from C57BL/6 mice purchased from Charles River Laboratories Japan, Inc.
Histological analysis
We used Oil Red O staining to evaluate liver fat deposition in paraffin-embedded liver tissue specimens.The ImageJ software (NIH) image software program was used to quantify the Oil Red O-stained areas in 8 microscopic fields at 400-fold magnification.
Immunoblot analyses
Proteins extracted from liver tissue were resolvedviapolyacrylamide gel electrophoresis, and the separated proteins in the gels were transferred to nitrocellulose membranes.The membranes were then probed with primary antibodies against SIRT1 (Merck, Tokyo, Japan), α-tubulin (Santa Cruz Biotechnology, Inc., TX, United States), phospho-AMP-activated protein kinase (AMPK), and total AMPK (Cell Signaling Technology Japan, K.K., Tokyo, Japan).The membranes were then incubated with corresponding horseradish peroxidase-conjugated secondary antibodies.The immunoreactive proteins were assessed with an LAS-4000 Image analyzer (FUJIFILM Holdings Corporation, Tokyo, Japan), and densitometry was performed using NIH.
Quantitative reverse transcription-polymerase chain reaction analysis
An RNAiso Plus kit (Takara Bio Inc., Shiga, Japan) was used for total RNA isolation.An Improm-II Reverse Transcription System (Promega Japan, Tokyo, Japan) was used for reverse transcription of isolated RNA into cDNA.cDNA samples (1 μg) were subjected to reverse transcription-polymerase chain reaction (RT-PCR) with a PCR Kit (TaKaRa) or to quantitative PCR with an Applied Biosystems ViiATM7 Real-Time PCR System (Life Technologies Japan, Ltd., Tokyo, Japan) and PowerUpTMSYBRTMGreen Master Mix (Fisher Scientific International, Inc., Pittsburgh, PA, United States).The specific primer sequences are listed in Table 1.The target mRNA expression levels were assessed relative to mouse β-actin mRNA (control gene) levels.
Statistical analysis
All data are presented as the mean ± SD.Multiple comparisons were performed with analysis of variance followed by post hoc tests, as appropriate.Pvalues of less than 0.05 were considered to indicate statistical significance.
RESULTS
Ipragliflozin reduced hepatic lipid accumulation regardless of body weight changes in ob/ob mice
All mice showed sensitive reactions, normal movement, normal appetite, normal stool, and stable breathing at the start of the experiment.The mice did not show any adverse events during the experiment, and no modifications of the experimental protocols were necessary.
We first tested whether ipragliflozin improved liver steatosis inob/obmice.There were no significant changes in body weight in either the 3 mg/kg or the 10 mg/kg ipragliflozin-treatedob/obmice compared with the untreatedob/obmice after 4 wk of treatment (Figure 1A).In addition, ipragliflozin did not significantly change the ratio of liver weight to body weight at the end of the experimental period (Figure 1B).On the other hand, we found that the livers of the 10 mg/kg ipragliflozin-treatedob/obmice had significantly lower Oil Red O-stained areas than those of the untreatedob/obmice (Figure 2).These results indicated that ipragliflozin improved liver steatosis irrespective of body weight changes.
Ipragliflozin increased hepatic SIRT1 protein expression levels in ob/ob mice
To elucidate the mechanism by which ipragliflozin improved liver steatosis inob/obmice, we examined the protein expression levels of hepatic SIRT1.Interestingly, compared with no treatment, ipragliflozin treatment significantly increased hepatic SIRT1 protein expression levels by approximately 2-fold inob/obmice (Figure 3).These results suggested that ipragliflozin upregulated the protein expression of SIRT1 in the livers ofob/obmice.
Ipragliflozin-mediated attenuation of liver steatosis in ob/ob mice was associated with SIRT1 signaling
Based on the abovementioned effect of ipragliflozin on the hepatic expression of SIRT1 protein, we analyzed the role of the SIRT1-PGC-1α/PPARα-FGF21 pathway in our mouse model.Specifically, we examined the mRNA expression levels of SIRT1, PGC-1α, PPARα, and FGF21 in the liver.Consistent with the findings regarding hepatic SIRT1 protein expression, we found that hepatic SIRT1 mRNA expression was significantly higher in 10 mg/kg ipragliflozin-treatedob/obmice than in untreatedob/obcontrol mice (Figure 4A).Moreover, we found that liver PGC-1α, PPARα, and FGF21 mRNA expression was significantly higher in ipragliflozin-treatedob/obmice than in untreatedob/obcontrol mice (Figure 4B-D).On the other hand, the hepatic mRNA levels of fatty acid synthase (FAS) and acetyl-CoA carboxylase (ACC), key regulators ofde novohepatic lipogenesis, did not significantly differ between the ipragliflozin-treatedob/obmice and the untreatedob/obcontrol mice (Figure 4E and F).These results indicated that the ameliorative effects of ipragliflozin on liver steatosis were possibly mediated by the SIRT1-PGC-1α/PPARα-FGF21 pathway.
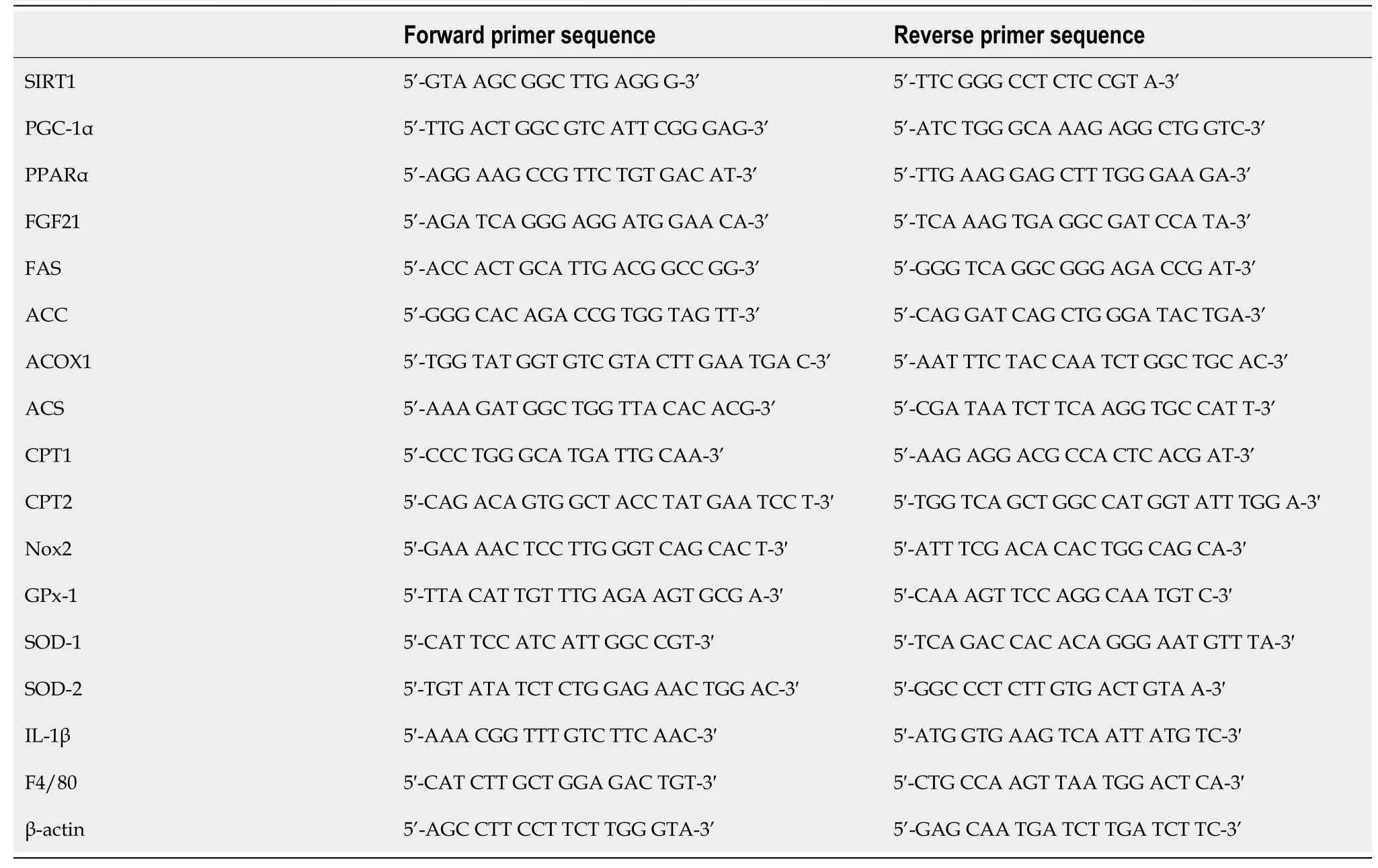
Table 1 Sequences of the primers used for reverse transcription-polymerase chain reaction
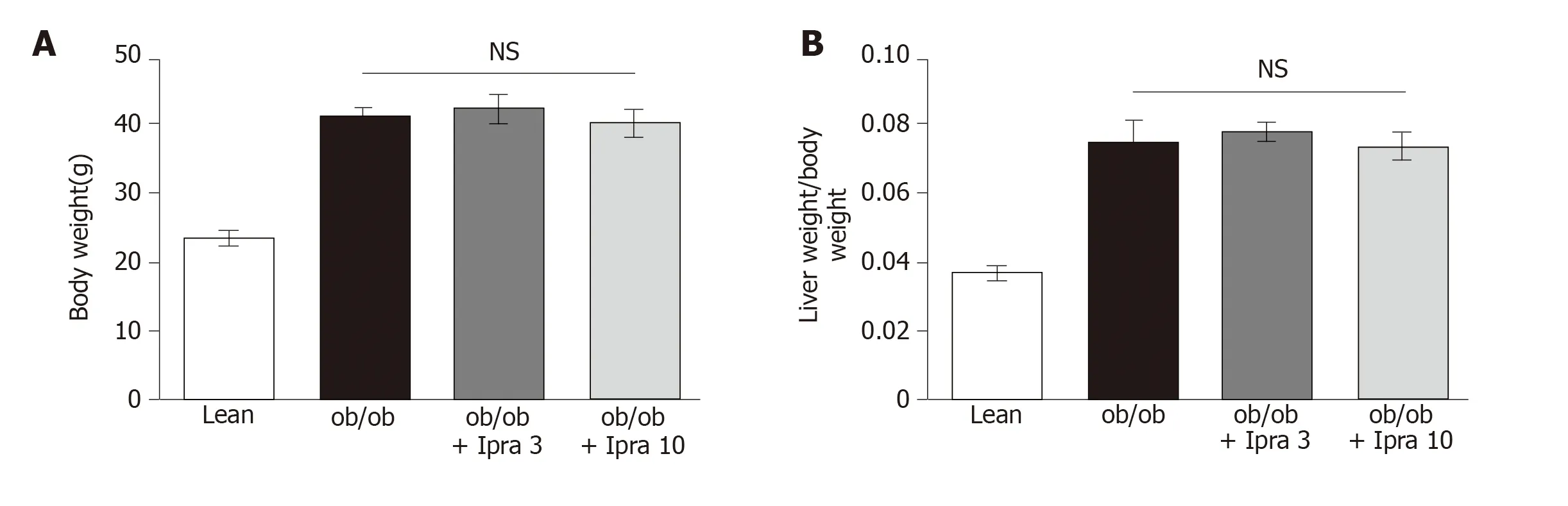
Figure 1 Body weights and liver-to-body weight ratios of obese mice treated with or without ipragliflozin and their lean littermates.
Ipragliflozin increased the mRNA expression levels of β-oxidation-related enzymes in ob/ob mice
Furthermore, we analyzed the mRNA expression levels of β-oxidation-related enzymes in our mouse model.We found that the hepatic mRNA expression levels of acyl-CoA oxidase 1 (ACOX1), acyl-CoA synthetase (ACS), carnitine palmitoyltransferase (CPT) 1, and CPT2 were significantly higher in 10 mg/kg ipragliflozin-treatedob/obmice than in untreatedob/obcontrol mice (Figure 5A-D).These results suggested that ipragliflozin increased both peroxisomal and mitochondrial β-oxidation inob/obmice.
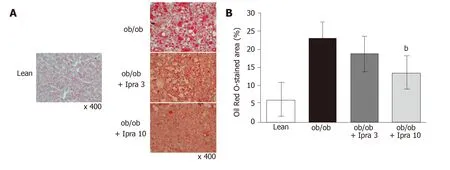
Figure 2 Evaluation of liver histology in obese mice treated with or without ipragliflozin and their lean littermates.

Figure 3 Hepatic sirtuin 1 protein expression in obese mice treated with or without ipragliflozin and their lean littermates.
Ipragliflozin decreased the mRNA expression levels of interleukin-1β but had no appreciable effects on those of oxidative stress-related genes or macrophage marker in ob/ob mice
We also analyzed oxidative stress, inflammatory cytokine levels, and macrophage infiltration in our mouse model.Treatment with 3 mg/kg ipragliflozin significantly increased the hepatic mRNA levels of NADPH oxidase 2 (Nox2), but treatment with 10 mg/kg ipragliflozin did not (Figure 5E).The hepatic mRNA levels of glutathione peroxidase 1, superoxide dismutase (SOD)-1, or SOD-2, key regulators of oxidative stress, did not significantly differ between ipragliflozin-treatedob/obmice and untreatedob/obcontrol mice (Figure 5F-H).Ipragliflozin decreased the mRNA expression levels of interleukin-1β (IL-1β) inob/obmice (Figure 5I).However, the hepatic mRNA expression levels of F4/80 were unchanged by ipragliflozin treatment (Figure 5J).
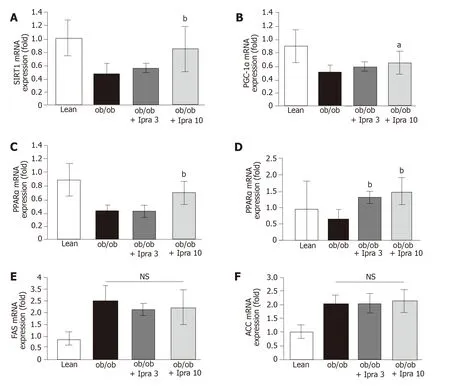
Figure 4 Hepatic mRNA expression of genes related to sirtuin 1 signaling in obese mice treated with or without ipragliflozin and their lean littermates.
SGLT2 was expressed in the kidneys but not in the liver
We next investigated why hepatic SIRT1 signaling was increased in ipragliflozintreated mice.We hypothesized that ipragliflozin increased hepatic SIRT1 signaling by directly inhibiting SGLT2 in the mouse liver.Therefore, we tested whether SGLT2 was expressed in the livers of mice.RT-PCR revealed that SGLT2 was expressed in mouse kidneys but not in mouse livers (Figure 6A).
Ipragliflozin increased AMPK activation in the whole liver
We further assessed the effects of ipragliflozin treatment on the activation of AMPK, a major metabolic energy sensor and master regulator of metabolic homeostatic processes, including SIRT1 signaling[23].Interestingly, ipragliflozin significantly increased the activation of AMPK in whole livers obtained from mice in the treated groups (Figure 6B and C).
DISCUSSION

Figure 5 Hepatic mRNA expression of genes related to β-oxidation, oxidative stress, inflammatory cytokine, and macrophage marker in obese mice treated with or without ipragliflozin and their lean littermates.
In our study, the SGLT2 inhibitor ipragliflozin ameliorated hepatic lipid accumulation in a manner associated with hepatic SIRT1 signaling in an experimental obese mouse model.It was unlikely that the inhibitory effect of ipragliflozin on liver steatosis was mediated by a decrease in body weight because ipragliflozin did not significantly affect body weight in the mouse model.Some SGLT2 inhibitors (empagliflozin, dapagliflozin, and canagliflozin) are generally reported to cause weight loss in obese mice and rats[24-26].However, consistent with previous reports[7,8], our results showed that ipragliflozin did not significantly alter body weight in obese mice (Figure 1A).It is possible that there are pharmacologic differences among SGLT2 inhibitors that cause them to have different effects on body weight.Further studies are required to confirm this hypothesis.
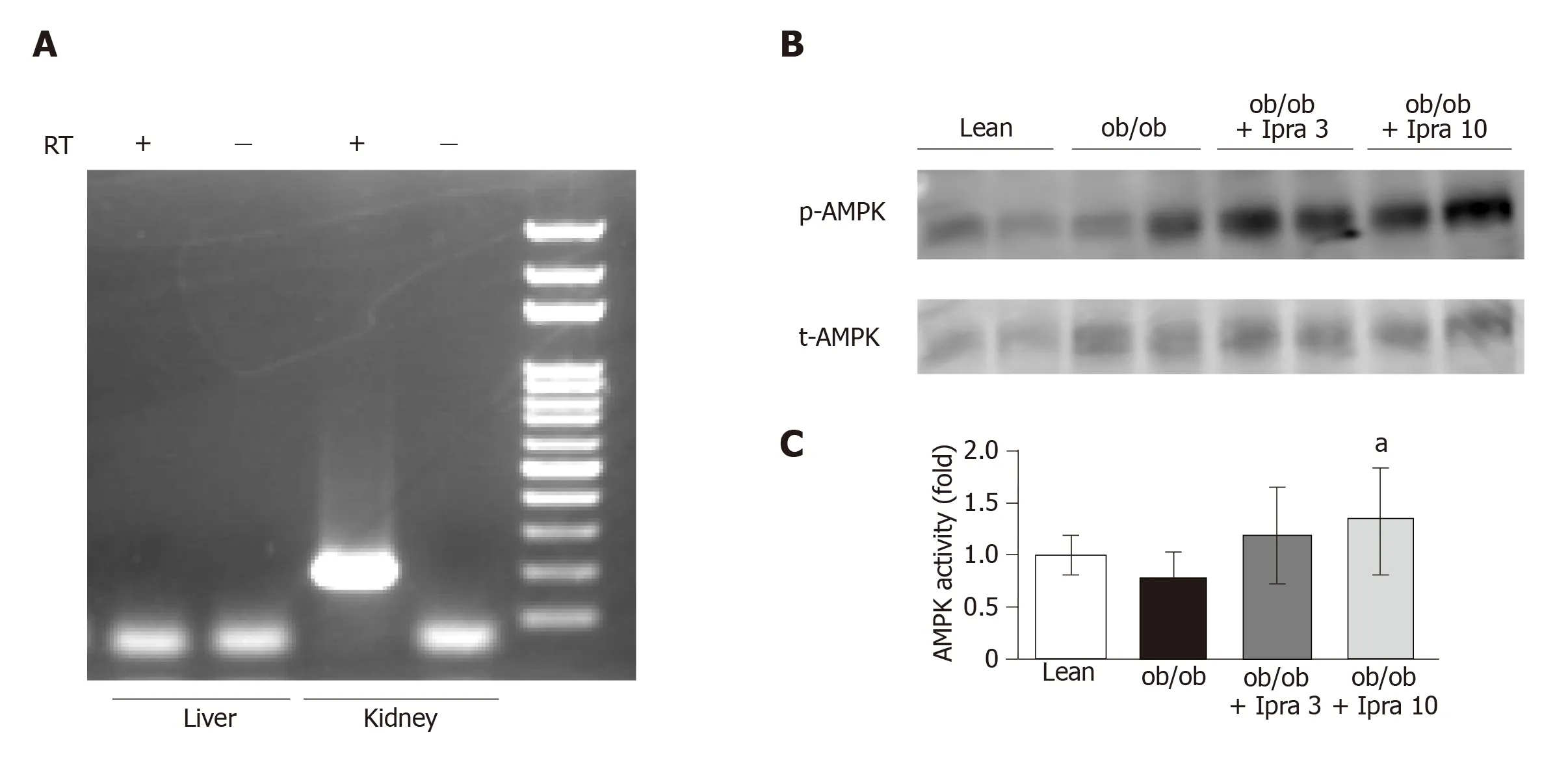
Figure 6 Hepatic and renal mRNA expression of sodium glucose cotransporter 2 and phosphorylation of AMP-activated protein kinase in whole livers of obese mice treated with or without ipragliflozin and their lean littermates.
The ameliorative effect of ipragliflozin on fatty liver in our experimental obese mouse model may have involved upregulation of the SIRT1 protein and subsequent enhancement of hepatic SIRT1 signaling.This hypothetical mechanism is supported by the following evidence: 1) hepatic SIRT1 protein expression levels inob/obmice were significantly increased by ipragliflozin treatment, and 2) activation of the SIRT1-PGC-1α/PPARα-FGF21 pathway was observed by mRNA expression analysis after ipragliflozin treatment.Ipragliflozin-mediated activation of the SIRT1-PGC-1α/PPARα-FGF21 pathway might result in promotion of mitochondrial fatty acid βoxidation and could account for the attenuation of liver steatosis inob/obmice.This mechanism is supported by a prior study demonstrating that ipragliflozin increases the hepatic mRNA expression levels of PPARα, a marker of lipid outflow, in Amylin liver NASH model mice[7].Moreover, the SGLT2 inhibitor empagliflozin has been reported to increase the hepatic mRNA levels of PGC-1α and FGF21 in mice with highfat-diet-induced obesity[24].However, a recent clinical study on NAFLD patients with T2DM reported that treatment with the SGLT2 inhibitor dapagliflozin decreased plasma FGF21 levels, while treatment with a combination of dapagliflozin and omega-3 carboxylic acids did not[27].FGF21 contributes to the regulation of mitochondrial activity and lipolysis in white adipose tissue[23,28]and increases fatty acid oxidation in the liver[22].Therefore, an increase in FGF21 in the liver following therapy with ipragliflozin may promote fat utilization.On the other hand, ipragliflozin did not change the hepatic mRNA expression levels of FAS and ACC, markers of lipid inflow, probably because it promoted hepatic fatty acid oxidation without suppressingde novohepatic lipogenesis inob/obmice.However, a previous report showed that the expression levels of FAS and ACC, which are upregulated in C57BL/6J wild-type mice fed a high-fat diet, are significantly suppressed by ipragliflozin[8].The exact reason for the inconsistency among these findings is unknown but might be related to the differences between genetically engineered and wild-type mice or the differences between the ipragliflozin administration methods used (dietary supplementationvsdrinking water supplementation).
Ipragliflozin did not significantly change the expression levels of oxidative stressrelated genes, with the exception of Nox2, the mRNA expression levels of which were altered in 3 mg/d ipragliflozin-treated livers; these findings are contradictory to the findings of a previous study[29].In addition, ipragliflozin did not significantly change macrophage infiltration based on the F4/80 mRNA expression data.In contrast, ipragliflozin significantly decreased IL-1β mRNA expression levels in the liver, consistent with the findings of a previous study[29].However, the inhibitory effect of ipragliflozin on IL-1β mRNA expression was relatively small; thus, ipragliflozin might have no appreciable effects on oxidative stress.The exact causes of the discrepancy between our results and previous results regarding hepatic oxidative stress are unknown; however, differences in the mice and/or experimental protocols used might have affected the results.
To our knowledge, this is the first report to show that the therapeutic effects of the SGLT2 inhibitor ipragliflozin are associated with the hepatic expression of the SIRT1 protein.Our findings are supported by a recent report to show that a decrease in renal SIRT1 protein expression was rescued by treatment with the SGLT2 inhibitor canagliflozin indb/dbmice[30].However, because SGLT2 is expressed in the kidneys but has not been reported to be expressed in the liver, further studies are needed to elucidate whether the effects of ipragliflozin on the liver are direct or indirect.Because SIRT1 is an energy-sensing molecule responsible for the promotion of healthy longevity mediated by caloric restriction[14], it is possible that temporary calorie loss due to the urinary glucose excretion caused by ipragliflozin may stimulate hepatic SIRT1.Similar conclusions were reached by Kimet al[31].In addition, AMPK enhances SIRT1 activity by increasing cellular NAD+ levels[32], and the interplay between SIRT1 and AMPK is suggested to be reciprocal[33].Several SGLT2 inhibitors, including canagliflozin, dapagliflozin, and empagliflozin, activate AMPK in HEK-293 cells, and canagliflozin activates AMPK in mouse liversin vivo[34].Such findings are consistent with our finding that ipragliflozin significantly enhanced AMPK activation in our mouse model.The activation of hepatic SIRT1 might have been partly due to the activation of AMPK in our mouse model.Further investigation is needed to elucidate the mechanism by which hepatic SIRT1 signaling is activated after treatment with the SGLT2 inhibitor ipragliflozin.
In a previous study, the hepatic mRNA and protein expression levels of not only SIRT1 but also SIRT3, SIRT5, and SIRT6 were found to be lower in a human NAFLD group than in a control group[35].Our findings from the comparison between the lean mouse group and the untreatedob/obcontrol mouse group regarding SIRT1 mRNA and protein expression are consistent with these results[35].The previous finding that SIRT1 activators inhibit the expression of lipogenic genes such as FAS and ACC[36,37]and similar findings that FAS and ACC expressions are increased while hepatic SIRT1 expression is repressed in the human NAFLD group[35]are also consistent with our data.Interestingly, the expression of SIRT4 has been found to be upregulated in humans with NAFLD compared with controls[35].SIRT4 mediates fatty acid oxidation in liver cells[38]and inhibits the interaction of SIRT1 and PPARα to decrease fatty acid oxidation[38].In our study, the mRNA expression levels of genes related to fatty acid oxidation, ACOX1, CPT1, ACS, and CPT2, were lower in untreatedob/obcontrol mice than in lean mice.These results suggest that the expression of SIRT4 might have been higher in untreatedob/obcontrol mice than in lean mice in our study.
The limitations of our study are that only one mouse model and only one SGLT2 inhibitor were used.The mechanisms of the effects of SGLT2 inhibitors on NAFLD should be verified in the future using several animal models of NAFLD and additional SGLT2 inhibitors.
In conclusion, this study suggests that the liver steatosis-attenuating effects of ipragliflozin inob/obmice may be mediated partly by hepatic SIRT1 signaling, possibly through the PGC-1α/PPARα-FGF21 pathway.Because SGLT2 inhibitors are widely used in clinical practice and are characterized by good safety and tolerability profiles, treatment with these inhibitors may be an effective therapeutic strategy for patients with liver steatosis induced by T2DM.
ARTICLE HIGHLIGHTS
Research background
The sodium glucose cotransporter 2 (SGLT2) inhibitor ipragliflozin has been reported to improve liver steatosis in animal models and clinical studies.However, the mechanisms by which SGLT2 inhibitors improve liver steatosis are not fully understood.To our knowledge, this is the first report to show that the therapeutic effects of the SGLT2 inhibitor ipragliflozin are associated with activation of sirtuin 1(SIRT1) signaling in the liver.
Research motivation
SGLT2 inhibitors are reportedly effective in fatty liver model mice as well as human nonalcoholic fatty liver disease patients.The mechanisms still need to be elucidated.Evaluating the mechanisms further may help identify molecules related to ameliorating fatty liver, allowing us to develop novel therapeutic strategies for fatty liver in the future.
Research objectives
The main objectives were to investigate the ameliorative effects of ipragliflozin on liver steatosis and the mechanisms of these effects in obese mice.Another objective was to evaluate the effect of ipragliflozin on β-oxidation, oxidative stress, inflammatory cytokine, and macrophage infiltration in the liver.Our study confirms the ameliorative effects of SGLT2 inhibitors on liver steatosis and the previously proposed mechanisms, and proposes a new mechanism, which can promote further research.
Research methods
Obese (ob/ob) mice and their littermates received a normal chow diet or a normal chow diet plus 2 doses of ipragliflozin for 4 weeks.We examined lipid accumulation, βoxidation, oxidative stress, inflammatory cytokine, and macrophage infiltration in the liver.Ob/obmice were suitable for this experiment as they developed fatty liver even when they received normal chow.In addition, we used two control mouse groups,ob/obcontrol mice that received ipragliflozin andob/oblittermates.In particular, SIRT1 signaling in the liver as a new candidate mechanism by which SGLT2 inhibitors improve liver steatosis was also assessed.
Research results
Amelioration of hepatic lipid accumulation by SGLT2 inhibitors was confirmed in our obese mouse model with ipragliflozin.Ipragliflozin-induced SIRT1 upregulation and SIRT1 signaling, which we propose might be involved in the mechanism by which ipragliflozin induces improvement of liver steatosis.The hypothesis should be further verified with different SGLT2 inhibitors in additional models and human samples.The observed effects of ipragliflozin on oxidative stress and macrophage infiltration, which were inconsistent with previous studies in the liver, need to be further evaluated.
Research conclusions
The new findings in our study are that SIRT1 signaling may be involved in the mechanism of ipragliflozin-induced improvement of liver steatosis inob/obmice.Thus, our study offers a new mechanism of ipragliflozin-induced improvement of liver steatosis.To be more specific, our proposed theory (hypothesis) is that activation of SIRT1 signaling due to ipragliflozin may ameliorate liver steatosis inob/obmice.This hypothesis and new phenomena were confirmed in our obese mouse model.In summary, the liver steatosis-attenuating effects of ipragliflozin inob/obmice may be mediated partly by hepatic SIRT1 signaling, possibly through the PGC-1α/PPARα-FGF21 pathway.The original insights into our results are that temporary calorie loss due to urinary glucose excretion caused by ipragliflozin may stimulate hepatic SIRT1, which might also be partly due to the activation of phospho-AMP-activated protein kinase in our mouse model.Our study provides additional evidence of SGLT2 inhibitor-induced improvement of liver steatosis; thus, we think that SGLT2 inhibitors are likely to be beneficial in diabetes mellitus patients with fatty liver in clinical practice.
Research perspectives
We take particular note of the role of SIRT1 as it plays important roles in controlling energy homeostasis and longevity in mammals and because the regulation of SIRT1 expression affects fatty liver.Thus, new target molecules that may be involved in amelioration of liver steatosis by SGLT2 inhibitors may be associated with energy homeostasis and longevity.We propose that future research should confirm our hypothesis using different animal models and human samples with different SGLT2 inhibitors or by confirming that upregulation or downregulation of SIRT1 signaling by the different methods used in our model or previous studies alters hepatic lipid accumulation.Genetically engineered mice, such as SIRT1 knockout mice, may be one of the best ways to confirm our results, and could be used to evaluate ipragliflozininduced improvement of liver steatosis.
 World Journal of Hepatology2020年7期
World Journal of Hepatology2020年7期
- World Journal of Hepatology的其它文章
- Oxidative stress in alcohol-related liver disease
- Anti-inflammatory and anti-oxidant effects of aloe vera in rats with non-alcoholic steatohepatitis
- Non-alcoholic steatohepatitis and the risk of myocardial infarction: A population-based national study
- Effect of zinc treatment on clinical outcomes in patients with liver cirrhosis: A systematic review and meta-analysis
- Diagnosis and management of hepatic artery in-stent restenosis after liver transplantation by optical coherence tomography: A case report
- Is right lobe liver graft without main right hepatic vein suitable for living donor liver transplantation?