
病毒调控应激颗粒形成的策略及应激颗粒对病毒复制的影响
2021-01-26 02:54郭鑫徐胜奎
南京农业大学学报 2021年1期
郭鑫,徐胜奎
(中国农业大学动物医学院,北京 100193)
哺乳动物细胞在外界环境压力的作用下会发生一系列应激反应,其中翻译起始复合物可聚集形成应激颗粒(stress granule,SG),阻碍细胞内mRNA的正常翻译[1]。病毒在细胞内寄生,完全依赖于宿主细胞的翻译系统合成病毒蛋白,完成自身生命周期。SG的形成对病毒的感染与复制具有重要调控作用。研究发现,从简单的单股正链RNA病毒到复杂的双链DNA病毒都可以调控宿主细胞SG的形成[2]。阐明SG与病毒感染之间的相互作用关系对于进一步了解病毒的致病机制具有重要意义。
病毒感染破坏细胞正常的生理功能和稳态,对宿主细胞也是一种应激。细胞可通过产生SG来下调整体的翻译效率,其一方面用于能量储存,另一方面则影响病毒蛋白的表达从而抑制病毒的复制。为了逃避宿主细胞对病毒的清除作用,病毒也进化出一系列措施抑制SG组装,发挥免疫逃逸的功能[3]。不同病毒的基因组、结构、大小及复制方式等各不相同,调控SG形成的策略也多种多样。病毒通过抑制细胞SG的形成,维持病毒蛋白的高效表达与基因组的有效复制,促进自身的感染[4]。一般认为,SG的形成影响宿主细胞的翻译效率,进而影响病毒的复制[5]。但是,也有研究表明SG的产生会抑制干扰素表达,有利于病毒的免疫逃逸[6]。由此可见,SG对不同病毒发挥的作用不尽相同。SG与病毒之间的相互影响关系可归纳为:宿主细胞感知病毒感染并诱导产生SG,同时病毒进化出多种方式干扰细胞SG的形成;此外,病毒还可利用SG来促进自身的复制。
1 SG的形成机制
SG的形成方式包括经典型(即eIF2α磷酸化依赖型)及非经典型(eIF2α磷酸化非依赖型)2种,其主要成分包括翻译停滞的mRNA、40S核糖体及多种宿主蛋白(如TIA-1、TIAR、Sam68、G3BP1和eIF等)[7]。在哺乳动物细胞中,真核起始因子2α(eukaryotic initiation factor 2 α,eIF2α)可被细胞内蛋白激酶R(protein kinase R,PKR)、血红素调节抑制激酶(heme-regulated inhibitor kinase,HRI)、一般性调控阻遏蛋白激酶2(general control non-derepressible 2,GCN2)及PKR样内质网激酶(PKR-like endoplasmic reticulum kinase,PERK)4种激酶磷酸化[8-9]。磷酸化的eIF2α影响eIF2-GTP-tRNAMet三聚体形成,阻碍翻译起始复合物组装,促进细胞内核糖核蛋白(RNP)聚集形成SG[10]。根据上述研究结果,笔者绘制了SG的形成机制示意图(图1)。
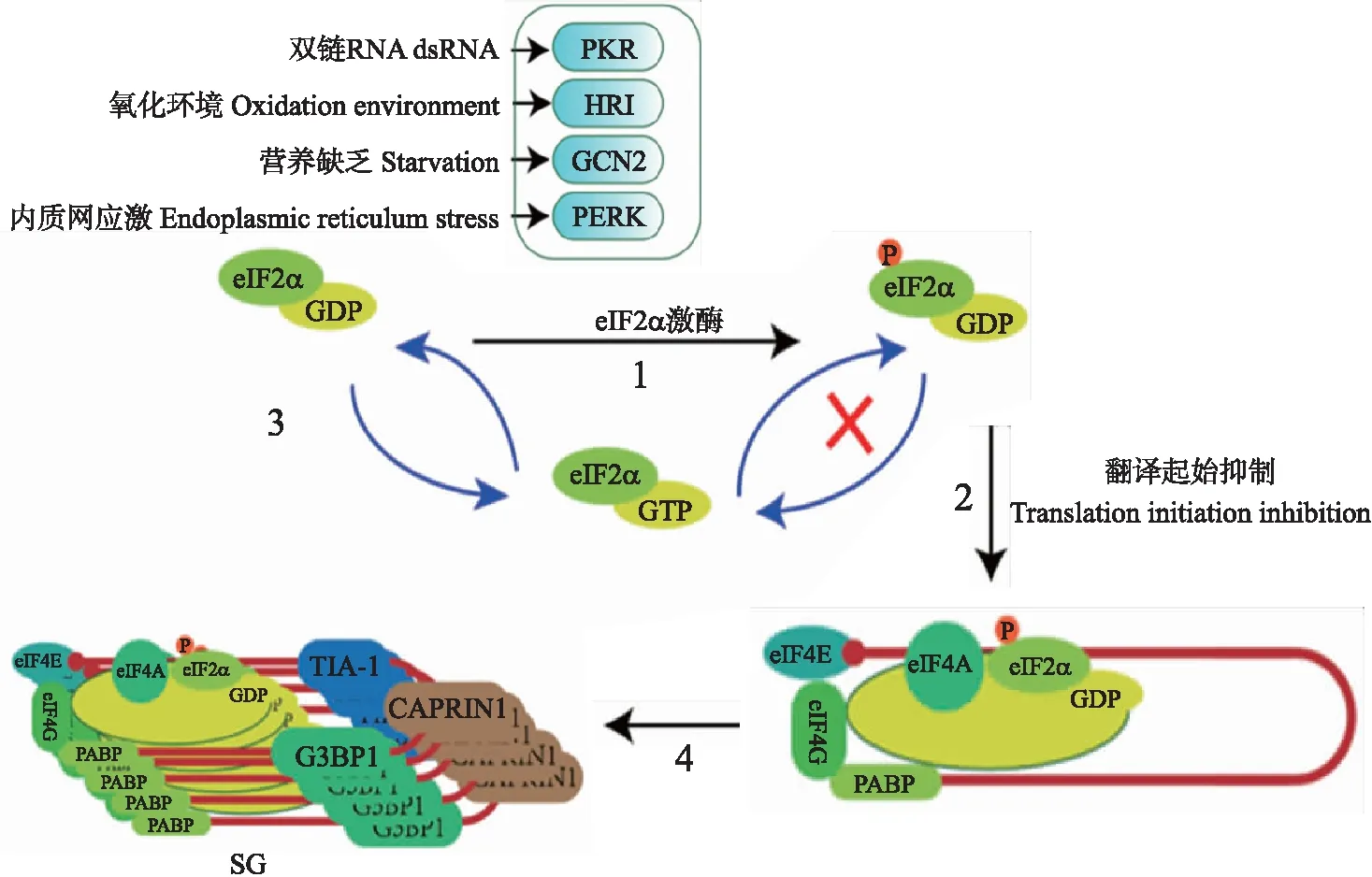
图1 经典型应激颗粒(SG)的形成机制Fig.1 Typical stress granule(SG)formation mechanism1. 细胞内有4种 eIF2α(真核起始因子2α)激酶:蛋白激酶R(PKR)、血红素调节抑制激酶(HRI)、一般性调控阻遏蛋白激酶2(GCN2)及PKR样内质网激酶(PERK)。面临不同的压力时,激酶的活化方式各异:病毒感染过程中产生的dsRNA是激活PKR的重要原因;细胞内产生的过氧化物以及热应激可使HRI活化;在细胞内营养不足时,GCN2被活化,使细胞保持较低水平的生命状态;病毒感染细胞后,病毒蛋白在内质网内大量合成,未正确折叠的蛋白质激活PERK。2. 当eIF2α被磷酸化后,eIF2α-GDP与eIF2α-GTP之间的循环发生障碍,细胞内无法生成足够的eIF2α-GTP,抑制细胞内的正常翻译。3. 未发生磷酸化修饰的eIF2α-GDP与eIF2α-GTP之间可进行正常交换。4. RNA结合蛋白(如G3BP1,TIA-1)与翻译停滞的mRNA结合形成复合物,RNA结合蛋白之间相互作用,使mRNA结合蛋白等复合物聚集形成SG。1. Four kinds of eIF2α(eukaryotic initiation factor 2α)kinases exist in host cell,including protein kinase R(PKR),heme-regulated inhibitor kinase(HRI),general control non-derepressible 2(GCN2)and PKR-like endoplasmic reticulum kinase(PERK). Kinases are activated in different ways in response to different stresses:dsRNA formed during virus infection may bind to and subsequently activate PKR;HRI could be activated by intracellular hyperoxide and hot stress;GCN2 could be activated under starvation environment to keep cells in a low state of life;excessive viral proteins synthesized in ER activate PERK in virus-infected cells. 2. The cycle between eIF2α-GDP and eIF2α-GTP is disrupted when eIF2α is phosphorylated,and the insufficiency of eIF2α-GTP inhibits normal translation. 3. The exchange between eIF2-GDP and eIF2-GTP occurs normally without phosphorylation. 4. RNA-binding proteins(such as G3BP1 and TIA-1)form complex together with untranslated mRNA and the interaction between RNA-binding proteins promote the assembly of SG.
在正常状态下,eIF2α的磷酸化水平由eIF2α激酶和磷酸酶协调控制并保持动态平衡。细胞内蛋白磷酸酶1(protein phosphatase 1,PP1)与PP2介导eIF2α去磷酸化。PP2在细胞内稳定表达,而PP1的表达量易受外界因素的调节[11-12]。PP1催化亚基PP1c通过与不同的调节亚基结合,靶向于不同的底物发挥作用,如PP1c与细胞内调节亚基生长停滞和DNA损伤可诱导蛋白34(growth arrest and DNA damage-inducible protein 34,GADD34)相互作用引导PP1作用于eIF2α使其去磷酸化[13]。低水平的磷酸化eIF2α不会造成RNP的聚集形成SG。因此,PP1c与GADD34的表达量可作为评估eIF2α去磷酸化酶活性的指标。
2 病毒刺激SG形成的方式
eIF2α磷酸化是SG形成的基础,其激酶的活化方式各异:PKR识别细胞浆内dsRNA并被活化;HRI可被细胞浆中活性氧激活;GCN2在氨基酸等营养物质缺乏的情况下发生活化;PERK可被内质网腔内大量错误折叠的蛋白质激活(图1)。在4种eIF2α激酶中,PKR和PERK的活化与病毒的感染密切相关。第1,RNA病毒进入宿主细胞后在细胞浆内进行复制形成复制中间体dsRNA,PKR作为模式识别受体首先识别并结合dsRNA从而引起自身寡聚化,寡聚化的PKR导致自身磷酸化,引起构象发生改变并暴露出eIF2α结合位点,随后PKR与eIF2α结合并使其磷酸化[14]。猪瘟病毒(Classical swine fever virus,CSFV)基因组为单股正链RNA,CSFV感染细胞后会促进PKR磷酸化,同时造成eIF2α的磷酸化;然而灭活的CSFV并不能活化PKR,也不会使eIF2α发生磷酸化,说明PKR的活化与病毒复制过程中形成的dsRNA密切相关[15]。同属于黄病毒科的寨卡病毒(Zika virus,ZIKV)与登革热病毒(Dengue virus,DENV)感染细胞后,同样会激活PKR并使eIF2α发生磷酸化,进而抑制细胞的翻译效率[14]。第2,囊膜病毒感染细胞后,需要合成大量的病毒囊膜糖蛋白完成自身的组装。内质网是病毒糖蛋白合成加工的主要场所,当内质网内蛋白合成过多过快时,造成内质网内未正确折叠蛋白的积累,激活位于内质网的激酶PERK,减缓蛋白的翻译速率,维持细胞稳态[9]。猪繁殖与呼吸综合征病毒(Porcine reproductive and respiratory syndrome virus,PRRSV)感染细胞后,病毒蛋白在内质网大量合成,激活细胞内质网压力感受器PERK使eIF2α发生磷酸化,进而刺激细胞产生SG[16]。马立克氏病病毒(Marek’s disease virus,MDV)为疱疹病毒,在病毒装配过程中需要合成大量的病毒囊膜蛋白,该病毒感染会激活细胞内质网未折叠蛋白反应[17]。病毒感染细胞后破坏细胞的稳态,除PKR与PERK之外,病毒也会激活细胞内其他eIF2α激酶,如辛德毕斯病毒(Sindbis virus,SV)感染细胞后可通过其病毒RNA与GCN2结合,直接激活GCN2,进而引起eIF2α的磷酸化[18]。综上,机体可从多个方面感知病毒,抑制其在宿主细胞的翻译,抵抗病毒感染。
除经典方式外,SG也可以eIF2α非依赖型的方式形成[19-20]。
3 病毒抑制SG形成的方式
SG最重要的功能是抑制宿主细胞的翻译,而病毒蛋白的表达及子代病毒的产生完全依赖于宿主的翻译系统,因此SG可作为宿主的天然免疫方式之一影响病毒的复制。病毒为了逃避宿主的清除作用,进化出多种策略抑制SG的形成[21-22]。多数SG的形成是eIF2α磷酸化依赖型,因此病毒可以从抑制eIF2α激酶的活化、促进eIF2α的去磷酸化以及直接破坏SG组成3个方面抑制SG的形成。笔者根据文献报道绘制了病毒调控SG形成策略示意图(图2)。
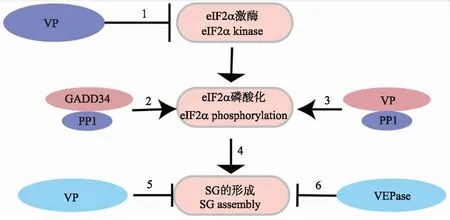
图2 病毒抑制SG形成的策略Fig.2 Virus strategies inhibiting SG assembly1. 病毒编码的蛋白可直接抑制eIF2α激酶的活化;2. 病毒感染刺激GADD34(调节亚基生长停滞与DNA损伤可诱导蛋白34)表达量提高,增强eIF2α的去磷酸化;3. 病毒蛋白(VP)可与蛋白磷酸酶(PP1)相互作用使eIF2α去磷酸化;4. eIF2α磷酸化促进SG的形成;5. VP可与SG组分相互作用,干扰SG的组装;6. 病毒编码的蛋白酶(VEPase)切割SG组份,抑制SG的产生。1. Virus-encoded proteins could inhibit the activation of eIF2α kinases directly;2. Virus infection elevates GADD34(growth arrest and DNA damage-inducible protein 34)production and subsequently enhance the dephosphorylation of eIF2α;3. Virus proteins(VP)may interact with protein phosphatase 1(PP1)to dephosphorylate eIF2α;4. eIF2α phosphorylation promote SG formation;5. Virus proteins may interact with SG components and disrupt SG assembly;6. Virus-encoded proteinase(VEPase)could cleave SG components and inhibit SG formation.
3.1 抑制eIF2α激酶的活化
eIF2α的磷酸化酶主要包括PKR、HRI、GCN2及PERK。病毒感染过程中可激活eIF2α激酶使其磷酸化,抑制eIF2α激酶的活化从而抑制SG的产生是病毒发挥作用的最直接方式。口蹄疫病毒(Foot and mouth disease virus,FMDV)感染PK-15细胞后激活细胞的天然免疫系统,诱导干扰素及干扰素刺激基因(如PKR)的表达[23]。Li等[24]报道FMDV感染PK-15后,尽管PKR基因的转录水平上升,但是病毒编码的3Cpro蛋白可通过溶酶体途径降解PKR,抑制eIF2α磷酸化和SG形成,从而促进病毒的复制。尽管流感病毒(Avain influenza virus,AIV)并不能在蛋白水平降解PKR,但有研究表明AIV编码的NS1蛋白N端结构域能与宿主细胞PKR相互作用,抑制PKR的活化,从而保证病毒的毒力与传播能力[25]。与RNA病毒不同,DNA病毒在复制过程中并不会产生dsRNA,但是其转录形成的RNA会形成二级结构激活PKR,因此DNA病毒也会进化出相应的策略抑制PKR的活化,如卡波济肉瘤相关疱疹病毒(Kaposi’s sarcoma-associated herpesvirus,KSHV)编码的ORF57蛋白可与PKR结合而破坏PKR与dsRNA之间的相互作用,抑制PKR自身的磷酸化及SG的形成[26]。人巨细胞病毒(Human cytomegalovirus,HCMV)感染后产生的dsRNA可作为PKR的激活剂,但是HCMV编码的pTRS1蛋白可与PKR直接作用而抑制dsRNA诱导的PKR活化,间接抑制SG形成[14]。值得注意的是,HCMV编码的pIRS1蛋白也会与HRI相互作用而抑制HRI的活化[14],表明病毒已经进化出多种策略抑制eIF2α激酶的活化。
除PKR外,病毒感染也可影响PERK的活化。多种病毒感染后可激活PERK,促进细胞内eIF2α磷酸化抑制宿主翻译,维持细胞稳态[16]。为了抑制eIF2α的磷酸化以维持病毒蛋白的翻译,单纯疱疹病毒(Herpes simplex virus,HSV)编码的gB蛋白可以特异性抑制PERK活化[27]。目前关于病毒抑制PERK活化的报道还很少,病毒调控SG的机制还需进一步研究。
3.2 促进eIF2α去磷酸化
当细胞内eIF2α磷酸化水平过高时,细胞内的负反馈机制将发挥作用,使eIF2α发生去磷酸化[28]。eIF2α磷酸化造成细胞内广泛的基因表达抑制,但是激活转录因子4(activating transcription factor 4,ATF4)与C/EBP同源蛋白(C/EBP-homologous protein,CHOP)的基因表达量却升高,ATF4与CHOP协同作用上调下游蛋白如转录因子ATF3及GADD34的表达[29]。GADD34作为PP1的调节亚基,它与PP1共同作用于eIF2α使其去磷酸化,维持细胞内eIF2α正常的磷酸化水平。GADD34的表达量易受环境的调控,因此病毒可通过提高GADD34的表达量使eIF2α磷酸化水平保持在较低水平,抑制SG的产生。基孔肯雅病毒(Chikungunya virus,CHIKV)感染细胞后,其在复制过程中产生的复制中间体dsRNA激活eIF2α的激酶PKR;同时该病毒可通过诱导GADD34表达增强细胞的去磷酸化作用,维持细胞的翻译水平[30]。病毒感染宿主后引发2个相互拮抗的反应,一方面病毒感染过程中激活PKR,使eIF2α发生磷酸化,促进SG的产生;另一方面,eIF2α的磷酸化诱导GADD34表达量升高,使eIF2α发生去磷酸化,这是病毒与宿主细胞相互博弈的结果。
3.3 直接破坏SG组成
病毒也可以直接作用于SG的主要组成成分,使其降解,或者阻碍SG的组装,进而发挥抑制SG形成的作用。黄病毒、小RNA病毒、杯状病毒等单股正链RNA病毒进入细胞后翻译成大的前体蛋白质,并在自身表达的蛋白酶作用下切割成熟。病毒表达的蛋白酶一方面可以水解自身的前体蛋白,同时还可以作用于细胞内的蛋白,发挥其免疫逃逸的作用。FMDV只含有1个开放阅读框,其编码的前导蛋白酶(leader protease,Lpro)可以直接切割SG组分即G3BP1与G3BP2,从而抑制SG的形成[31]。猫杯状病毒(Feline calicivirus,FCV)感染细胞后会使eIF2α发生磷酸化,但是并不会刺激细胞产生SG[32]。此外,FCV感染还会抑制亚砷酸钠诱导的SG,多点突变试验结果表明,FCV感染过程中产生的病毒蛋白酶NS6(Pro)会切割SG主要成分G3BP1,以此破坏SG的组装[32]。CHIKV尽管不会破坏SG的组成成分,但是其编码的非结构蛋白nsp3含有SH3结构域结合区域,可招募G3BP1从而干扰SG的组装[33]。西尼罗病毒(West Nile virus,WNV)的负链RNA 3′端的颈环结构可与SG组成成分TIA-1/TIAR相互作用,阻碍SG的组装[34]。
值得注意的是,有些病毒可通过多种方式抑制SG的形成。如DENV不仅抑制eIF2α非依赖型SG形成,同时还可抑制eIF2α磷酸化依赖型的SG的形成[35]。传染性支气管炎病毒(Infectious bronchitis virus,IBV)可以通过非结构蛋白nsp2抑制PKR的活化从上游抑制eIF2α的磷酸化,还可以上调GADD34的表达水平和提高PP1的去磷酸化酶活性,抑制eIF2α的磷酸化并影响SG形成[36]。HSV可通过表面糖蛋白gB抑制PERK的活化和抑制eIF2α的磷酸化[27];同时HSV蛋白ICP34.5可以通过与PP1相互作用而促进eIF2α的去磷酸化,抑制SG的形成[37]。以上结果表明,病毒可以在SG形成的任何阶段对其进行调控。
4 SG对病毒复制的影响
细胞受到应激(如病毒感染)时,往往通过维持自身最基本的代谢进行能量储存,以抵抗恶劣的生存环境。越来越多的证据表明,病毒感染过程中可通过抑制宿主细胞内SG的形成,促进病毒自身蛋白的翻译。SG的抗病毒作用主要表现为以下几种形式:部分病毒的翻译严格依赖于细胞中的翻译起始因子如40S亚基与eIF4G,SG中滞留的翻译起始因子不利于病毒蛋白的翻译[38]。SG的主要组成成分TIA-1与TIAR通过与某些病毒3′端的颈环结构相互作用来调控病毒的复制,当TIA-1与TIAR滞留于SG后,病毒的复制水平将受到影响[34];丙肝病毒(Hepatitis C virus,HCV)在感染过程中会将SG的组成成分G3BP1、ATX2、PABP1等招募到病毒复制复合体周围辅助病毒的复制,当G3BP1等滞留于SG中时,影响病毒复制复合体的组装[39];Onomoto等[40]报道,滞留于SG中的RNA识别受体RIG-I可被SG中的dsRNA活化进而激活细胞的天然免疫应答;同样地,破坏SG形成会影响HSV感染过程中PKR的活化[41]。因此,SG为病原相关分子模式的识别提供平台,激活宿主细胞的天然免疫信号通路,有利于病原的清除。
尽管SG的形成可发挥抗病毒的作用,但也有研究报道SG可有效抑制细胞的天然免疫反应,有利于病毒感染。细胞中转录生成mRNA的翻译是帽子依赖型的,需要宿主内完整的翻译起始因子参与,而DENV与ZIKV等病毒含有内部核糖体进入位点,不依赖于帽子结构就可起始翻译[42]。SG中滞留的翻译起始因子会抑制抗病毒因子如干扰素的表达,然而病毒的蛋白表达并不受影响,在此情况下SG的形成有利于病毒的感染。
5 总结与展望
SG的形成是一个动态的过程。由于SG含有大量mRNA及多种RNA结合蛋白,组成成分复杂,各成分的功能及其对于SG组装的作用还需要进一步阐明。深入研究SG的组装机制不仅有助于深入了解宿主的抗病毒机制,还可为抗病毒药物的研发提供新思路。
SG影响病毒在细胞内的复制,通过人为调控SG的形成可间接调控病毒的复制,在培养病毒及制作灭活苗时提高病毒滴度,降低生产成本。另外,在SG形成过程中,滞留于SG中的mRNA多为含有帽子结构的宿主细胞基因,拥有核糖体进入位点的病毒基因依然可以有效翻译,SG捕获mRNA是否具有选择性依然值得深入研究。病毒的致病性与调控SG形成的能力密切相关,利用基因编辑技术对病毒基因进行人为改造,调控SG的形成能力,降低病毒的致病能力,可作为弱毒疫苗的候选毒株,为疫苗研发提供思路。
猜你喜欢
中老年保健(2022年1期)2022-08-17
现代临床医学(2021年1期)2021-01-26
智慧健康(2020年20期)2020-12-05
小读者之友(2020年4期)2020-05-15
科学24小时(2018年1期)2018-01-10
现代养生·下半月(2016年6期)2016-10-21
医学研究杂志(2015年12期)2015-06-10
医学研究杂志(2015年12期)2015-06-10
癌变·畸变·突变(2015年3期)2015-02-27
癌变·畸变·突变(2015年3期)2015-02-27
