
POAG相关基因与中枢神经系统疾病的研究进展
2021-03-10 08:11王艾嘉
国际眼科杂志 2021年3期
王艾嘉,张 旭
0引言
青光眼是一种神经退行性疾病,其临床表现为渐进性视盘凹陷性萎缩和视野特征性缺损,是造成不可逆致盲的主要原因[1]。原发性开角型青光眼(primary open angle glaucoma,POAG)约占青光眼的60%~70%,其病因尚不完全明了,遗传可能是病因的重要因素。自遗传连锁分析和全基因组关联研究(genome-wide association study,GWAS)分别应用于POAG的研究以来,两种方法已鉴定了大量POAG相关基因和风险基因位点。与此同时,随着影像学和神经科学水平的提高,青光眼开始被认为是中枢神经系统(central nervous system,CNS)疾病的一种类型。研究表明,青光眼患者在视网膜神经节细胞(RGCs)至大脑枕叶视皮质的整个视觉通路上均出现病理改变,而晚期青光眼患者结构和功能的改变甚至延伸到了CNS的非视觉区域[2-3]。已知的神经退行性疾病包括肌萎缩侧索硬化症(ALS)、额颞叶痴呆(FTD)、阿尔茨海默病(AD)、亨廷顿病(HD)、帕金森氏病(PD)和脊髓小脑性共济失调(SCA)等。遗传是神经退行性疾病的重要病因,建立POAG相关基因与CNS疾病的联系将对“青光眼是一种CNS疾病”的观点提供重要依据。本文将介绍主要的POAG相关基因及其与CNS疾病之间的联系。
1 POAG的遗传连锁分析
1.1遗传连锁分析的应用概况遗传连锁分析是一种基于家系的,用于定位与相关家系中特定表型相关的染色体区域的方法,主要应用于孟德尔性状或遗传可能性高的性状。通过对POAG家系的遗传连锁分析人们确定了一系列基因位点并依次从GLC1A编码至GLC1P。该方法筛选出的POAG相关基因包括MYOC、CYP1B1、WDR36、OPTN、TBK1以及其它的一些候选基因。
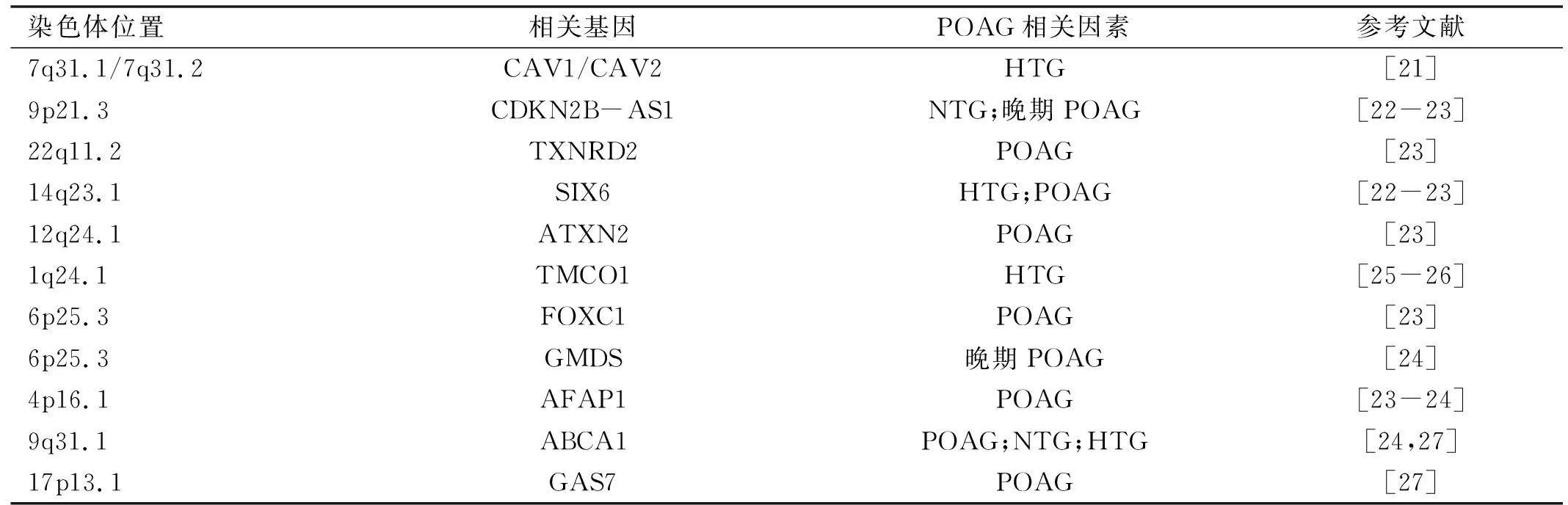
表1 通过GWAS确定的POAG相关基因
1.2遗传连锁分析确定的基因位点MYOC是Stone等[4]发现的第1个POAG致病基因,该基因位于连锁位点GLC1A。MYOC约85%的突变是位于第3外显子的错义突变。MYOC的突变主要与青少年型原发性开角型青光眼(juvenile open angle glaucoma,JOAG)有关[5]。而位于连锁位点GLC3A的CYP1B1则被Stoilov等[6]确定为是原发性先天性青光眼的致病基因。Kaur等[7]发现CYP1B1作为MYOC的修饰物可使MYOC与CYP1B1相互作用,但MYOC和CYP1B1的相互作用与POAG发病机制之间的联系仍知之甚少[8]。
WDR36是位于连锁位点GLC1G的POAG致病基因[9]。虽然在约6%的POAG患者中检测出了4个WDR36突变(N355S、A449T、R529Q和D658G)[9],但这些突变也存在于健康受试者中,这使得WDR36在POAG中的作用受到质疑。Liu等[10]在关于WDR36的Meta分析中也指出其对于POAG遗传易感性不起主要作用。因此关于WDR36在POAG发生中作用的重要程度仍存在争议。
OPTN最早由Sarfarazi等[11]定位于连锁位点GLC1E。该基因编码一种泛素结合的自噬受体蛋白Optineurin。OPTN突变已被发现与正常眼压性青光眼(normal tension glaucoma,NTG)有重要联系,目前已确定的OPTN突变中以E50K错义突变最为常见,该突变参与DNA结合和蛋白质二聚化,可对OPTN功能造成显性负效应[12]。E50K突变在NTG中的作用尚不完全清楚,Shim等[13]发现E50K突变通过改变 Bax通路和线粒体动力学引起线粒体降解和自噬体形成,这可能是其导致POAG的重要机制。TBK1位于连锁位点GLC1P,负责编码丝氨酸/苏氨酸激酶。TBK1基因的拷贝数变异(CNVs)被发现可以通过自噬失调导致早发性家族性NTG。OPTN与TBK1在多种生化途径中有密切联系,两者的相互作用可以激活细胞自噬,而过度表达E50K则可以增强OPTN与TBK1的相互作用[14]。OPTN或TBK1的突变被认为会导致自噬的异常激活以及不可逆转的RGCs破坏,这为青光眼发生提供了一种机制[15]。同时,OPTN和TBK1突变与CNS疾病之间的关联也被越来越多研究者提出。
1.3其它遗传连锁分析确定的基因位点NTF4位于连锁位点GLC1O。虽然Pasutto等[16]在1.7%的欧洲POAG患者中发现了7个NTF4杂合突变,但其它地区的研究均未重复这一结果。IL20RB位于连锁位点GLC1C,Keller等[17]在一个大型POAG家族中发现IL20RB的1个罕见T104M突变,研究表明T104M突变很可能通过影响IL-20信号通路导致家族性青光眼的发生[18]。ASB10位于连锁位点GLC1F,TNF-α和IL-1α上调小梁网细胞中ASB10的表达支持了ASB10在青光眼中的作用[19]。EFEMP1位于连锁位点GLC1H,EFEMP1功能障碍导致POAG的可能机制包括基底膜结构功能受损、内质网应激慢性激活导致的细胞死亡[20],且EFEMP1突变与视神经的视盘面积减少相关。
2 POAG的全基因组关联研究
2.1 GWAS的应用概况GWAS以流行病学为基础,用于评价某种疾病与人群中某种基因突变是否存在显著关联。近年来,GWAS在对复杂性疾病的研究中取得了显著进展,自该方法运用于POAG研究以来已经鉴定出数十个POAG相关基因,且检测出的基因数量仍在上升。
2.2由GWAS确定的风险基因位点Liu等[8]曾对GWAS确定的POAG相关基因进行系统的总结,表1列出了其中较为主要的基因,包括CAV1/CAV2[21]、CDKN2B-AS1[22-23]、SIX6[22-23]、TXNRD2[23]、ATXN2[23]、FOXC1[23]、GMDS[24]、TMCO1[25-26]、ABCA1[24,27]、AFAP1[23-24]、GAS7[27]。Youngblood等[28]通过对全球多地多种族的研究又新发现近30种POAG相关的单核苷酸多态性(single nucleotide polymorphism,SNP),标志着未来GWAS在POAG领域研究的潜力。同时ATXN2在一些CNS疾病中起关键作用,我们可借此建立青光眼与CNS疾病在遗传学层面的联系。
3 POAG相关基因与CNS疾病的联系
3.1 OPTN与TBK1
3.1.1肌萎缩侧索硬化症肌萎缩侧索硬化症(ALS)是一组由脊髓、脑干和大脑中的运动神经元进行性死亡引起的神经退行性疾病。高达10%的ALS患者具有ALS家族史,表明该病的遗传特征[29]。导致ALS的OPTN突变最早由Maruyama等[30]发现,研究表明ALS中的OPTN突变可能具有奠基者效应。在人类患者OPTN的N-末端区域共检测到5个错义突变,其中与ALS相关的R96L突变可干扰OPTN的寡聚状态或OPTN与其他结合体的相互作用,而包括E50K在内的其余4个突变则与POAG相关;OPTN的C-末端区域与ALS连锁的突变包括错义突变Q454E、E478G以及截断突变Q398X。数据表明这些突变可干扰或破坏OPTN UBAN结构域的功能,进而影响OPTN依赖的选择性自噬过程[31]。Minegishi等[29]也认为多数ALS相关的OPTN突变是因为缺乏泛素结合所需的C-末端区域,因此OPTN中的ALS突变似乎与泛素结合所支配的功能有关。McCauley等[32]研究则表明OPTN突变可能通过改变全身和神经炎性反应影响疾病进展,进一步拓展了OPTN突变导致ALS的机制研究。
值得注意的是,Swarup等[33]发现OPTN突变具有一定的细胞特异性。在细胞培养中,青光眼相关突变E50K、M98K会选择性诱导RGCs凋亡;而ALS相关突变E478G则诱导运动神经元样细胞死亡,但不诱导RGCs死亡。因此一般来说,OPTN的青光眼相关突变和ALS相关突变之间没有关联。但有1个罕见的插入突变(691_692InsAG)被发现同时与NTG[12]和ALS[34]相关。虽然其致病机制目前尚不清楚,但这提示POAG与ALS之间的联系可能更为紧密,即累及OPTN的同一突变可能导致青光眼和(或)ALS的发生。
Cirulli等[35]和Freischmidt等[36]研究已证明TBK1基因是ALS的致病基因。研究发现,TBK1基因的E696K错义突变和C-末端区域的p.690-713缺失突变可干扰和阻止OPTN与TBK1的相互作用,通过影响自噬或线粒体自噬参与ALS的发生。后续Freischmidt等[37]进一步提出ALS相关的TBK1错义突变可导致单倍体不足从而影响自噬功能,这一观点在de Majo等[38]研究中得到了证实。此外,TBK1还参与如微管动力学[39]、微管网络[40]等可能与ALS致病相关的细胞机制。
因此可以推测,无论是引起相关功能障碍的OPTN突变,还是影响TBK1与OPTN相互作用的TBK1突变,最终都将影响与神经退行性疾病高度相关的OPTN依赖的自噬与有丝分裂功能。
3.1.2额颞叶痴呆额颞叶痴呆(FTD)作为额颞叶变性(FTLD)的一种分型,是一种因额叶和(或)颞叶的退行性变导致的一组临床综合征。30%~50%的FTD患者有至少一位亲属出现类似病情[41],提示其高度的遗传性。OPTN突变在FTD中的作用目前尚存在争议,在一项对104例没有运动神经元受累的FTD患者中鉴定出4.8%的患者具有OPTN和TBK1突变[42],而另一项对371例FTD患者研究的试验则无法检测到OPTN突变[43]。尽管如此,数据显示OPTN突变仍然影响超过1%的FTD患者[42]。此外,Farhan等[44]在2个患有ALS和FTD的家族中均检测到OPTN的1个罕见的p.Met468Arg突变,并提出该突变在C9orf72病理扩增的前提下可进一步调整疾病表型,但该突变是否可以单独导致ALS或FTD尚不清楚。
Pottier等[42]研究确定了TBK1突变是FTLD-TDP(FTLD最常见的病理亚型)的常见原因,并发现导致OPTN和TBK1功能缺失的突变可通过影响蛋白质聚集影响OPTN/TBK1通路在CNS疾病(如FTLD和ALS)中的保护作用;与在ALS中的作用机制类似,TBK1的单倍体不足可能损害TBK1在自噬和神经炎症中的功能;同时TBK1的CCD2结构域缺失被发现会使TBK1与OPTN的相互作用受阻,导致ALS/FTD发生[35-36]。最近则有报道在行为变异型FTD患者中TBK1的p.lle334Thr突变通过神经炎症途径参与FTD-ALS谱系疾病的发生[45]。
由于FTD与ALS是具有共同潜在发病机制的疾病谱的一部分,OPTN与TBK1在两者中相近的致病机制并不令人十分惊讶。但3.1.1中POAG与ALS的联系提示青光眼也可能属于这个疾病谱,这将进一步佐证POAG与CNS疾病的关系。
3.1.3阿尔茨海默病和亨廷顿病阿尔茨海默病(AD)作为一种进行性神经退行性疾病,其主要病理特征是淀粉样斑块和神经原纤维缠结(NFT)。虽然关于POAG与AD之间的联系被多次提出[46-47],但OPTN与TBK1在AD中的发现仍十分有限。Liu等[48]在AD的NFT和营养不良性神经突中发现了Optineurin,并提出了OPTN参与神经变性和细胞死亡进程的假说,但目前仍缺少遗传学证据证明OPTN在AD中的作用。关于TBK1,Verheijen等[49]在包含1 253例早发型AD(EOAD)患者的欧洲人群中仅发现1例TBK1突变,他们认为这可能只是1个具有AD早期症状的非典型FTD患者,TBK1对于EOAD不起到主要作用。另一项研究中,1个具有AD表型的TBK1 p.D534H突变在1个晚发型AD(LOAD)家族中被发现,研究结果支持该患者AD的诊断[50]。尽管因为研究数据的缺乏,TBK1突变参与AD发生这一观点目前尚存在争议,但现有发现仍说明TBK1突变可能造成临床区分AD和FTD的困难,且由于仍然存在TBK1突变导致AD的可能性,故对具有AD表型的患者进行TBK1基因突变筛查仍是有必要的。
亨廷顿病(HD)是一种常染色体显性遗传的神经退行性疾病,该病致病基因的代谢产物为亨廷顿蛋白(Huntingtin,HTT)。Schwab等[51]研究发现HD中Optineurin存在于神经核和核周包裹体中,提出Optineurin与HTT的相互作用可能是HD的致病机制。Li等[52]进一步证明OPTN UBAN结构域的泛素结合能力是OPTN与HTT-polyQ(polyQ proteins derived from human Huntingtin)共定位的关键因素。但目前仍缺乏证据证明OPTN在HD中的遗传学作用。
3.1.4帕金森氏病流行病学已经证实了帕金森氏病(PD)与青光眼之间的联系。PD患者的POAG发病率明显上升,且更易表现为NTG[53],在PD患者视觉通路上也观察到了病理变化。而关于POAG与PD的遗传学联系近年来也有所发现,Lill等[54]进行的GWAS研究发现,OPTN的青光眼相关突变M98K被发现是PD的危险因素;Lamb等[55]在1例皮质基底节变性的患者中检测到了1个新的TBK1突变(p.E703X),并预测该突变会破坏OPTN/TBK1的相互作用并损害自噬功能,这表明TBK1突变可能是导致非典型PD的原因。尽管基于这些发现PD与青光眼有了一定的关联,但两者的联系仍需进一步探索。
3.2 ATXN2
3.2.1肌萎缩侧索硬化症正常情况下,ATXN2编码区含有1个由CAG重复序列构成的聚谷氨酰胺(PolyQ)结构域,该结构域包含22~23个谷氨酸,由核苷酸序列(CAG)8CAA(CAG)4CAA(CAG)8-9编码。PolyQ结构域的病理扩展可导致神经萎缩,影响神经元的投射。ATXN2的中等长度CAG扩展(Q27-33)[56]已被确定为ALS的危险因素,且ALS的患病风险随等位基因重复呈指数增加。Elden等[56]发现ATXN2的中等长度扩展在引起ALS的同时伴有TAR DNA结合蛋白(TDP-43)的出现,扩展的ATXN2更易与TDP-43结合并修饰TDP-43的毒性,以此参与疾病的发生。Farg等[57]则在ALS患者中发现ATXN2与FUS(fused in sarcoma)蛋白的共定位,并证明中等长度扩展的ATXN2可以增强两者的相互作用以及修饰FUS的病理作用。Lattante等[58]首次报道ATXN2中等长度扩展与家族性FTD-ALS显著相关,他们提出该扩展可以在存在C9orf72扩展的前提下充当FTD表型的强大修饰物,导致以FTD和ALS为特征的临床表现。van Blitterswijk等[59]在另一项试验中也提出ATXN2的中等长度扩展可能对扩展的C9orf72起修饰作用。另有研究在ALS和FTD表型的患者中观察到ATXN2中等长度扩展[44],间接支持了上述观点。但ATXN2扩展并不单独与FTD相关[59]。事实上,TDP-43与FUS是ALS的主要致病因素,C9orf72也是FTD的主要致病因素。因此可以推测,ATXN2在ALS-FTD系谱疾病中更多的是增强或修饰主要致病因素的作用,而非独立引起疾病发生。
3.2.2脊髓小脑性共济失调脊髓小脑性共济失调(SCA2)是一种常染色体显性遗传的神经退行性疾病,ATXN2中CAG序列的重复扩展已被证明为SCA2的致病原因[60]。但与ALS不同,导致SCA2的CAG扩展序列更长,CAG重复次数多达37~39个,且通常无CAA序列中断,即表现为单纯的CAG重复[61]。长扩展的ATXN2导致SCA2的机制包括参与RNA的加工、翻译和新陈代谢的调节,ATXN2扩展后获得新的毒性功能,使浦肯野细胞放电频率变得异常缓慢并最终导致细胞丢失[62]。Li等[63]则发现ATXN2在SCA2患者的脑组织和在有ATXN2扩展的ALS组织中双向转录,含有CUG序列重复扩增的反义转录产物ATXN2-AS具有神经毒性,研究表明ATXN2-AS可能有助于SCA2和ALS的发生。
此外,有研究在SCA2患者中发现PD的伴随,并指出伴随发生的PD与CAG扩展(Q33-43)相关[64]。Monte等[65]则证明了PD与CAG长扩展之间的确切联系,并指出CAG扩展内的CAA中断是目前研究最多的PD遗传因素。
ATXN2长扩展是SCA2以及PD的重要原因,相关发病机制的研究已较为成熟。结合3.2.1可以发现,目前在建立 POAG与CNS疾病的联系方面,ATXN2的作用十分有限。但ATXN2在CNS疾病发病机制中发挥着重要作用,作为青光眼与CNS疾病共同的危险因素,未来ATXN2在青光眼的研究中仍具有意义。
4总结
本文首先介绍了POAG主要的相关基因。遗传连锁分析和GWAS两种检测方法确定了大量POAG相关位点,极大推进了POAG病因的研究。以对疾病靶向干预从而实现精准医疗为目标,基于大数据的基因检测方法在未来势必还有更大的发展空间。此外本文重点对POAG相关基因与CNS疾病之间的关系进行了总结和思考,已知的CNS疾病在临床表现、病理学和遗传学等方面表现出许多相似之处,通过对POAG相关基因与CNS疾病之间联系的总结归纳可以发现POAG在许多方面,特别是遗传学方面,与CNS疾病都有着很高的相关和相似程度。这些发现均为“青光眼是一种CNS疾病”的观点提供了有力支撑。
目前,关于POAG及其与CNS疾病的联系仍存在许多疑问。如ATXN2在青光眼中的致病机制,ATXN2在CNS疾病中的参与程度,OPTN突变的细胞特异性以及OPTN和TBK1在AD等CNS疾病中的遗传学证据等问题均有待进一步探索。最后,如何继续在分子水平探寻POAG与CNS疾病之间的联系、深入了解青光眼患者CNS的改变机制以寻求对青光眼本质的正确认识将是今后青光眼领域相关研究的重点。
猜你喜欢
分子催化(2022年1期)2022-11-02
分析测试学报(2022年9期)2022-09-21
中国农业科学(2022年16期)2022-09-19
中老年保健(2022年3期)2022-08-24
散文诗(青年版)(2021年6期)2021-08-09
眼科学报(2021年6期)2021-07-18
健康之家(2019年3期)2019-12-14
汽车观察(2019年2期)2019-03-15
电脑知识与技术(2018年19期)2018-11-01
金桥(2018年9期)2018-09-25
