
Okur-Chung neurodevelopmental综合征1例并文献复习
2021-03-11 03:21王爱萍杨洋尹丽娟张红红许芳
中国生育健康杂志 2021年2期
王爱萍 杨洋 尹丽娟 张红红 许芳
Okur-Chung neurodevelopmental综合征是一种以特殊面容和多系统受累为主要表现的疾病,该病首次由Okur等报道,既往报道的病例中都存在有CSNK2A1基因的突变[1]。该基因被证实与神经发育障碍性疾病(neurodevelopmental disorders,NDD )有关[2]。由于缺乏确切的临床诊断指标,Okur-Chung neurodevelopmental综合征的诊断在没有基因诊断前较为困难。本文报道1例基因诊断的Okur-Chung neurodevelopmental综合征患者,并通过文献复习介绍此病的临床表现、诊断要点及治疗建议。
一、病例资料
患儿男,9岁5个月,主诉因生长迟缓9年余就诊。患儿出生后家长即发现患儿生长迟缓,较同年龄儿童慢,年生长速率不详,无慢性头痛、恶心、呕吐或视物模糊等,有明显智力、运动及语言发育落后,目前上小学,平素注意力不集中,多动,学习成绩差,饮食欠佳,睡眠可,二便可。出生史:G2P2,足月顺产,出生体重3.2 kg,出生时有轻度窒息抢救史,Apgar评分不详,姐姐15岁,身体健康。喂养史:生后母乳喂养,吃奶差,有反复吐奶,现普食,胃纳欠佳,体重增长缓慢。生长发育史:6月抬头,1岁会坐,不会爬,1岁8月会站,2岁会走,1岁出牙,2岁出齐,3岁会说话。既往史:平素有反复呼吸道感染,否认外伤史、过敏史、按时按序预防接种。家族史:父亲身高178 cm,母亲身高158 cm,遗传靶身高174.5 cm。父母为汉族,非近亲结婚,智力和面容正常。入院查体:体温36.5℃,心率92次/分,呼吸20次/分,血压85/55 mmHg。一般情况及反应可,神志清晰,无大理石皮肤、红斑、无皮肤干燥、无头皮黑色素沉着,颈抗(-),双侧瞳孔等大等圆,对光反射存,浅表淋巴结无肿大,全身无皮疹,甲状腺无明显肿大,咽(-)。三凹征(-)、呼吸平稳、双肺呼吸音粗,未闻及干湿罗音,心律齐,心音有力,各瓣膜听诊区未闻及病理性杂音,腹软,不胀,无压痛,肝脾未触及,肠鸣音正常,未见脐疝及腹股沟疝,四肢肢端暖,足跟CRT 1~2秒,四肢肌力、肌张力正常,膝反射正常,病理征阴性。专科情况:患儿面容特殊(图1),头颅小(头围49 cm),头发色泽正常,倒三角脸,前额扁平,发际线高,一字眉,大眼,外眦下斜,眼距窄,眼袋明显,间歇性内斜视,鼻梁宽,耳位低下,耳廓小,轻度听力丧失,薄上唇,舌头大,舌尖分叉,高腭弓,大上门牙,双手指短小,双手掌纹无异常,双足趾细长。身高115 cm(低于同种族、同年龄、同性别正常儿童平均身高3个标准差),体重21.5 kg(低于同种族、同年龄、同性别正常儿童平均体重2个标准差),阴茎3 cm.,双侧睾丸3 mL,未见阴毛生长。辅助检查:血常规正常;尿常规正常;肝功、肾功、心肌酶、电解质、葡萄糖检测正常;幽门螺旋杆菌抗体检测阴性;生长激素药物激发试验(左旋多巴+精氨酸):0 min 1.21 ng/mL,30 min 3.36 ng/mL,60 min 2.11 ng/mL,90 min 27.27 ng/mL,120 min 9.67ng/mL;25-羟基维生素D 37.39 nmol/L;肾上腺功能正常;甲状腺功能正常;胰岛素生长因子151.3 ng/mL(参考值88~452 ng/mL);甲胎蛋白、癌胚抗原正常;乙肝表面抗体阳性;微量元素正常。智力评估65分(智力低下)。心脏彩超检查心内结构未见异常。骨龄片(G-P图谱法):左手腕部骨化中心出现9颗,尺骨骨化中心出现,茎突不明显,籽骨未出现,骨龄评估为6岁6月(图2)。垂体MRI平扫+增强检查提示:蝶鞍无扩大,形态未见异常。垂体体积小,上缘稍凹陷,体积约为3.9 mm×5.7 mm×9.8 mm(上下径×前后径×左右径),神经垂体显示正常,平扫及增强未见异常信号。垂体柄稍向右偏,信号未见异常。双侧海绵窦、视交叉未见异常。双侧蝶窦粘膜增厚(图2)。在征求家长知情同意后,留取患儿及父母新鲜外周血样本送由北京迈基诺医学检验所应用安捷伦外显子芯片捕获及高通量测序方法检测医学全外显子筛查中包括的全部基因的外显子突变情况。患儿血样本在Okur-Chung neurodevelopmental综合征相关基因CSNK2A1外显子区域发现一处杂合突变点:c.479A > G(编码区第479号核苷酸由腺嘌呤变异为鸟嘌呤),导致氨基酸改变p.His160Arg(第160号氨基酸由组氨酸变异为精氨酸,为错义突变),遗传方式为常染色体显性遗传(AD),家系验证结果表明其父母此位点无突变(图3)。根据美国医学遗传学与基因组学学会(ACMG)新版指南[3]对变异进行分析:(1)PS2。经家系验证分析,受检人之父该位点无变异,受检人之母该位点无变异,此变异为自发突变;(2)PM2。在正常人群数据库中的频率为低频变异;(3)PP3。生物信息学蛋白功能[4]预测软件SIFT、PolyPhen_2、REVEL分别预测为有害、有害、有害;(4)HGMD数据库未有该位点的相关报道。综上所述考虑该变异初步判定为疑似致病性变异(likely pathogenic)。
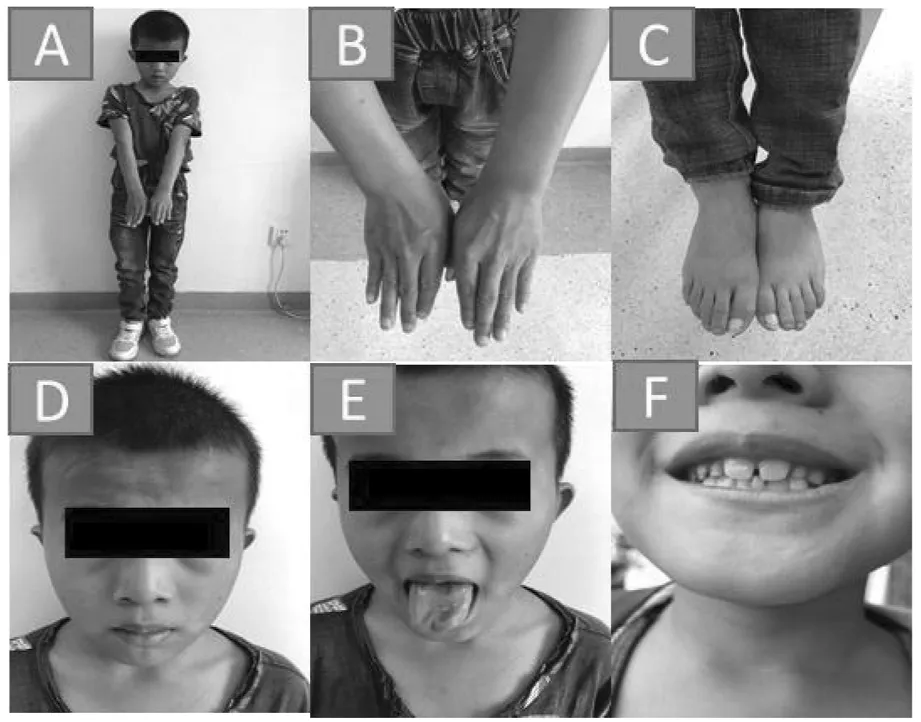
A.身材矮小 B.双手指短小 C.双足趾细长 D.特殊面容E.舌大、舌尖分叉 F.大上门牙图1 患儿照片

注:患儿存在CSNK2A1基因c.479A > G杂合变异(箭头示),患儿母亲和父亲该位点无变异(箭头示)图2 患儿骨龄(6岁6月)、垂体MRI(垂体体积小、垂体柄稍向右偏)
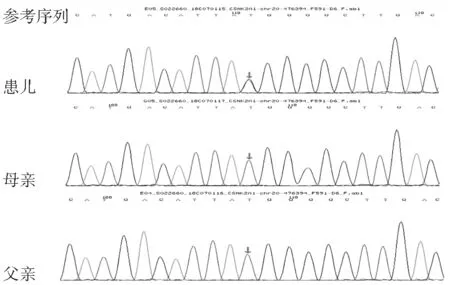
图3 Okur-Chung 综合征家系CSNK2A1基因测序结果
二、文献复习并总结
1.CSNK2A1基因与Okur-Chung neurodevelopmental综合征:既往报道证实,CSNK2A1基因位于常染色体,人类RNA序列分析显示CSNK2A1基因在人脑的各个部分、左心室、淋巴结、肾脏、脾脏和卵巢均有表达[5]。 CSNK2A1基因编码蛋白激酶Ⅱ(CK2)的α亚单位,是一种丝氨酸/苏氨酸激酶,包含两个催化亚基(α和α′)和两个调节亚基(β),在激酶反应过程中需以GTP或者ATP作为磷酸盐供体。它普遍存在于各种组织中,并且在细胞的生长、增殖和细胞凋亡中发挥着重要作用[6]。小鼠胚胎敲出CSNK2A1基因会导致神经管发育缺陷、心血管畸形、胎儿水肿和胚胎死亡[7]。既往在各类的癌症报道中CSNK2A1基因表达增强[8],而在T细胞性白血病中CSNK2A1基因表达缺失[9]。CK2还可以促进骨髓源性抑制细胞的生长,骨髓源性抑制细胞可以抑制适应性免疫应答和调节免疫反应[10]。这可以解释在Okur-Chung neurodevelopmental综合征患儿中均存在免疫缺陷。所有表达CSNK2A1的组织在Okur-Chung neurodevelopmental综合征患儿中都会有相应的临床特征。在Chiu等[11]对Okur-Chung neurodevelopmental综合征的研究中报道了8种CSNK2A1基因突变,其中6个新发突变,2个既往曾报道过的突变,包括病例1 c.593A>G,p.(Lys198Arg);病例2 c.79G>A,p.(Glu27Lys);病例3 c.1A>G,p.(Met1Val);病例4 c.692C>G,p.(Pro231Arg);病例5 c.218T>A,p.(Val173Glu);病例6 c.140G>A,p.(Arg47Gln);病例7 c.935G>A,p.(Arg312Gln);病例8 c.151A>C,p.(Ser51Arg)(表1),而本文报道的病例也为新生变异c.479A > G,p.His160Arg。虽然CK2在各种各样的细胞功能中发挥着重要作用,例如代谢调控、基因表达、细胞周期和细胞增殖,但是其确切的分子学机制仍然是不清楚的。在CSNK2A1同源基因敲出的小鼠模型中,尽管发生了胚胎死亡,但是也证实了CSNK2A1基因缺失与神经管发育缺陷、小头畸形、肢体发育迟缓和鳃弓不融合有关,这与Chiu等[11]报道病例的临床表型具有相似性,同时也提示在CSNK2A1基因变异的患儿中存在蛋白质功能的缺失。由此推测Okur-Chungneurodevelopmental综合征的发病机制是由于CSNK2A1基因变异导致了蛋白质功能的缺失所致。
2. Okur-Chung neurodevelopmental综合征的临床特征:Chiu等[11]对14例存在CSNK2A1基因突变患儿的临床特征进行了总结,具体如下。
(1)神经发育障碍。智力发育障碍是最主要的临床特征,14例病例中有13例存在智力发育障碍(92.9%),除了病例1,正常上学而且成绩处于中上水平。行为问题包括孤独症(3/14,21.4%),注意力缺陷多动障碍(4/14,28.6%),睡眠紊乱(8/14,57.1%),脾气暴躁(3/14,21.4%)。肌张力低下观察到的频数为9/14(64.3%),既往报道的MRI影像学检查发现的各种各样的异常(6/14,42.9%),例如巨脑回、继发大脑外侧裂扩大、胼胝体变薄、蚓发育不良、大脑皮质变薄和松果体囊肿。1例病例(7.1%)存在脑积水需要神经外科干预。2例病例(14.2%)存在共济失调,另外1例病例存在运动障碍。本文报道的病例存在明显的智力运动语言发育落后、注意力不集中、多动、学习成绩班级倒数第一,与既往报道病例一致。
(2)多器官系统受累。多器官系统受累,最常见的是生长发育迟缓和或矮身材(8/14例,57.1%)。28.6%(4/14)的患儿有皮肤的异常,例如大理石皮肤、帕尔默红斑、皮肤干燥以及头皮黑色素沉着。57.1%(8/14)的患儿存在消化系统受累,例如便秘、胃肠道动力不足和胃食管返流,其中2例病例(2/14例,14.3%)需要胃造口术。42.9%(6/14)的病例免疫系统受累:21.4%(3/14)的病例存在免疫功能异常,21.4%(3/14)的病例反复感染。其他问题包括脐疝(1/14,7.1%)和腹股沟疝(2/14,14.3%)。仅有1例病例存在肺发育不良、手指细长、唇粘连、间歇性内斜视、轻度的听力丧失、怕热、易疲劳、贫血、两排牙齿、虹膜缺损/瞳孔不等,牙龈肥大、结膜灰斑和肉碱缺乏。尽管既往报道CSNK2A1基因变异与癌症相关,但是14例患儿中没有1例存在恶性肿瘤。本文报道的病例存在生长发育迟缓、身材矮小,生后吃奶差,有反复吐奶,平素饮食欠佳,有反复呼吸道感染,轻度听力丧失,与既往报道病例一致,此外,本文报道的病例还发现患儿双手指短小、双足趾细长。
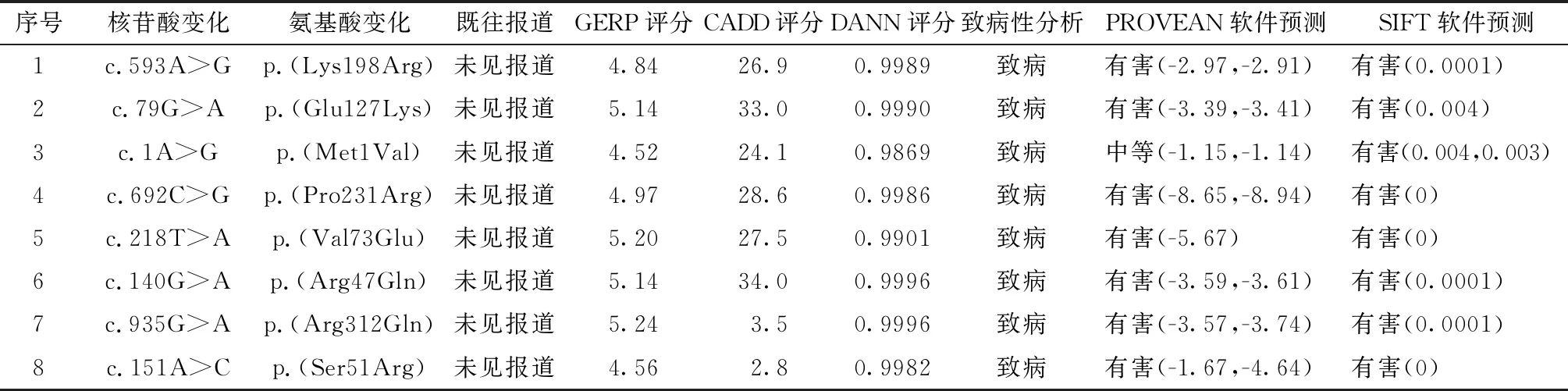
表1 Okur-Chung neurodevelopmenta综合征患者CSNK2A1基因突变类型
(3)特殊面容。最常见的面部特征包括小头畸形(8/14,57.1%)、圆脸(4/14,28.6%)、眼距增宽(3/14,21.4%)、部分上眼睑下垂(3/14,21.4%)、柳叶眉(4/14,28.6%)、内眦赘皮(4/14,28.6%)、鼻梁宽(6/14,42.9%)、耳位低(4/14,28.6%)、耳位低(4/14,28.6%)、耳廓异常(6/14,42.9%)。其他报道过的特征包括斜头畸形、扁平脸、杏仁眼、间歇性内斜视、虹膜缺损或者瞳孔不等、高腭弓、薄上唇、后颌畸形或小颌畸形、舌头大、两排牙齿和大上门牙。本文报道的病例小头畸形、鼻梁宽、耳位低、耳廓小、间隙内斜视、高腭弓、薄上唇、舌头大、大上门牙,与既往报道的病例一致,此外,本文报道的病例还发现患儿倒三角脸、前额扁平、发际线高、一字眉、大眼、外眦下斜、眼距窄、眼袋明显、舌尖分叉。
3. Okur-Chung neurodevelopmental综合征的治疗建议:
(1)临床遗传学家的评估。在诊断Okur-Chung neurodevelopmental综合征的时候临床遗传学家需回顾患儿的临床和分子学信息,并且做遗传学咨询。同时需注意这个综合征的预后,尤其要注意基因分析报告,是一个新发的致病变异,还是一个已经观察到的致病变异。
(2)整体神经发育评估及多学科训练。Okur-Chung neurodevelopmental综合征最主要的临床特征就是神经发育缺陷。早期干预中要结合整体神经发育评估,采取适当的物理治疗、作业治疗及远期语言训练治疗。对于那些怀疑有自闭症或者注意力缺陷多动障碍的患儿建议临床精神科医师或者儿科精神科医师做远期评估。当患儿年龄大于6岁时,需做智力评估同时给予适当的教育。
(3)生长、营养和骨骼发育发育的监测。每年定期监测生长发育和骨骼发育情况。因为CSNK2A1与生长发育异常有关,这些患儿具有发生胃肠道问题例如消化功能异常、胃食管反流而导致营养不良,这些问题不容忽视。如果检测到骨骼畸形建议完善影像学检查和咨询骨科医生。
(4)头围的测量。定期监测头围。60%的患儿存在小头畸形,头围的增加需注意影像学检查除外脑积水[11]。有异常神经系统表现的患儿如惊厥、共济失调和严重的肌张力低下需完善脑部影像学检查,有惊厥和运动异常的患儿还需完善脑电图检查。
(5)免疫功能评估。由于Okur-Chung neurodevelopmental综合征的患儿存在经常或严重的感染,需监测免疫球蛋白的基础水平,同时咨询免疫学家。
(6)此外,还需注意有无脐疝和腹股沟疝的发生,必要时咨询儿外科医生明确是否需要手术。
总之,由于目前报道病例数过少,Okur-Chung neurodevelopmental综合征的临床表型仍然是不明确的,因此也不能得出Okur-Chung neurodevelopmental综合征的诊断标准,临床上没有一个症状和体征具有绝对的诊断意义,因此,基因分析发现致病突变仍然是确诊该病的重要手段。
猜你喜欢
英语文摘(2021年11期)2021-12-31
支部建设(2020年15期)2020-07-08
儿童故事画报(2020年12期)2020-06-23
家庭医学·下半月(2019年6期)2019-08-16
小天使·二年级语数英综合(2017年3期)2017-04-01
琴童(2016年9期)2016-05-14
奥秘(2015年4期)2015-09-10
百科知识(2015年18期)2015-09-10
小星星·阅读100分(高年级)(2015年4期)2015-05-26
中学生物学(2008年12期)2008-12-27
