
通过泰乐内酯羟基的选择性氧化改进泰地罗新合成工艺
2021-06-03 07:58赵伯龙曾淑云
中国兽药杂志 2021年5期
于 荣,赵伯龙,郭 佳,曾淑云
(宁夏泰益欣生物科技有限公司, 银川 750205)
泰地罗新(图1)是一种新型泰乐菌素衍生类广谱抗菌药,对一些革兰氏阳性和革兰氏阴性细菌均具有抗菌活性[1-3]。目前有报道的合成泰地罗新的主要方法,均为对泰乐菌素(图2)的水解产物C20,23上的取代基进行修饰,得到20,23-哌啶基-5-O-碳酶氨糖-泰勒内酯[4~6],即泰地罗新。其中C-20羟基的修饰为合成泰地罗新反应中最关键的步骤。但目前有文献报道的方法,大部分是对C-20羟基进行碘代后,与哌啶进行亲核取代。
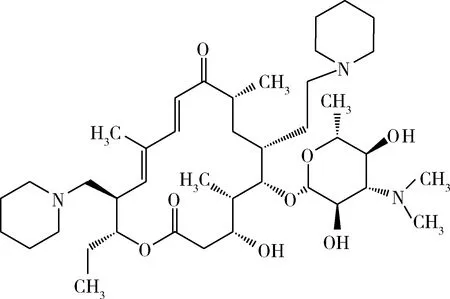
图1 泰地罗新Fig 1 Tildipirosin
采用这种路线合成泰地罗新主要面临两个难点问题:碘化剂价格昂贵,碘化反应合成效率低,都会造成泰地罗新产品的成本居高不下。无论是采用碘、碘盐还是NIS之类的有机碘化物作为碘代剂,成本都是工业化过程中绕不开的难题。由于泰地罗新是用于规模化家畜养殖过程中的呼吸道疾病的预防与治疗,预期使用规模极大。如果药品价格过高,将直接影响产品的市场前景。因此,需要一种新的合成路线,取代碘化物的使用,降低泰地罗新的合成成本。
也有报道采用硼氢化钠还原C-23位羰基为羟基[7],同时将两个羟基进行卤代。但羟基卤代过程如使用三苯基膦法[8],生成的三苯氧磷难以分离,且污染环境。之后使用对甲苯磺酰氯进行卤代[9],生成的对甲苯磺酸酯与卤代物极性相似,难以判断反应得率也很难分离产物。

图3 20-羟基-23羰基-5-O-碳酶氨糖泰乐内酯Fig 3 5-O-Mycarose tylosin
研究同样以修饰C-20羟基入手,采用TEMPO选择性氧化法[10],利用C-20羟基是泰乐内酯上唯一伯羟基进行选择性氧化,将C-20羟基氧化为羰基,再与哌啶反应,合成泰地罗新。
1 材料与设备
表1与表2的内容为本研究所使用的主要原料与设备仪器。
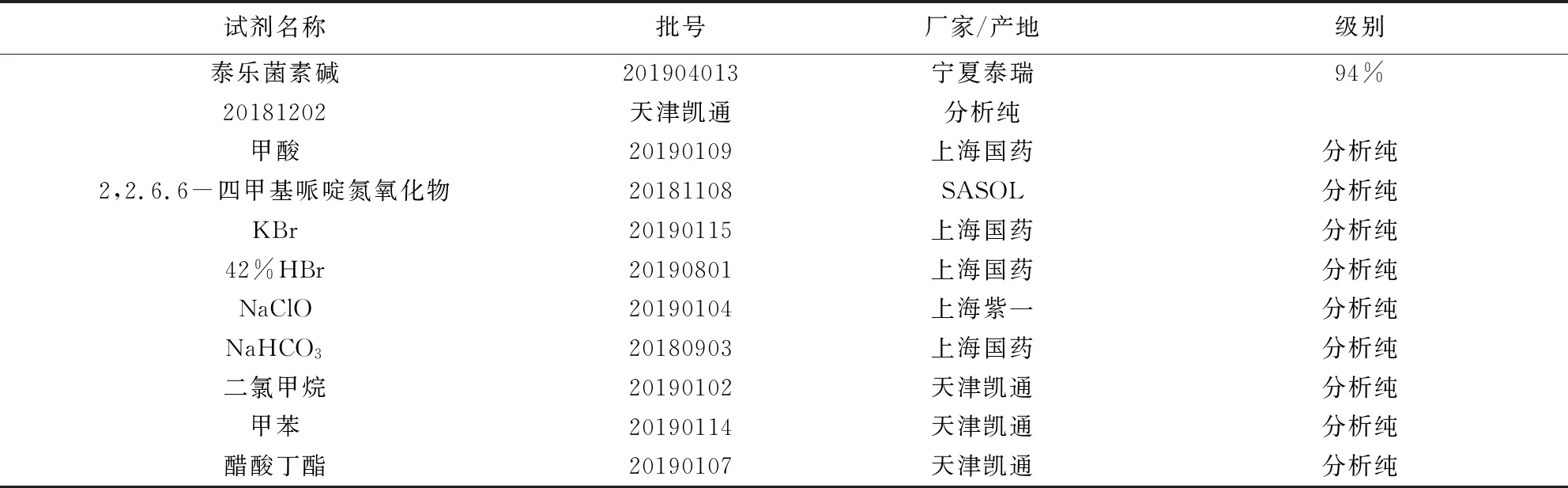
表1 所用主要试剂列表Tab 1 List the main reagents used

表2 所用主要仪器设备列表Tab 2 The main equipment list
2 实验方法
实验为避免C-23位羰基对TEMPO氧化的影响,先将其进行氨基取代,然后对取代物进行水解,使C-20羟基暴露,再进行TEMPO氧化,得到C-20羰基化的泰乐内酯,再第二次使用氨基取代,得到泰地罗新。
2.1 C-23的Wallach加成反应 取18.32 g(0.02 mol)泰乐菌素碱,溶于200 ml甲苯中。氮气保护下升温至45 ℃,加入1.89 g(0.022 mol)哌啶,混合均匀后,升温至70 ℃。缓慢滴加1.11 g(0.024 mol)甲酸,滴加完毕后保温反应120 min,降温至室温,得到23-哌啶基-泰乐菌素碱的甲苯溶液(图4)。
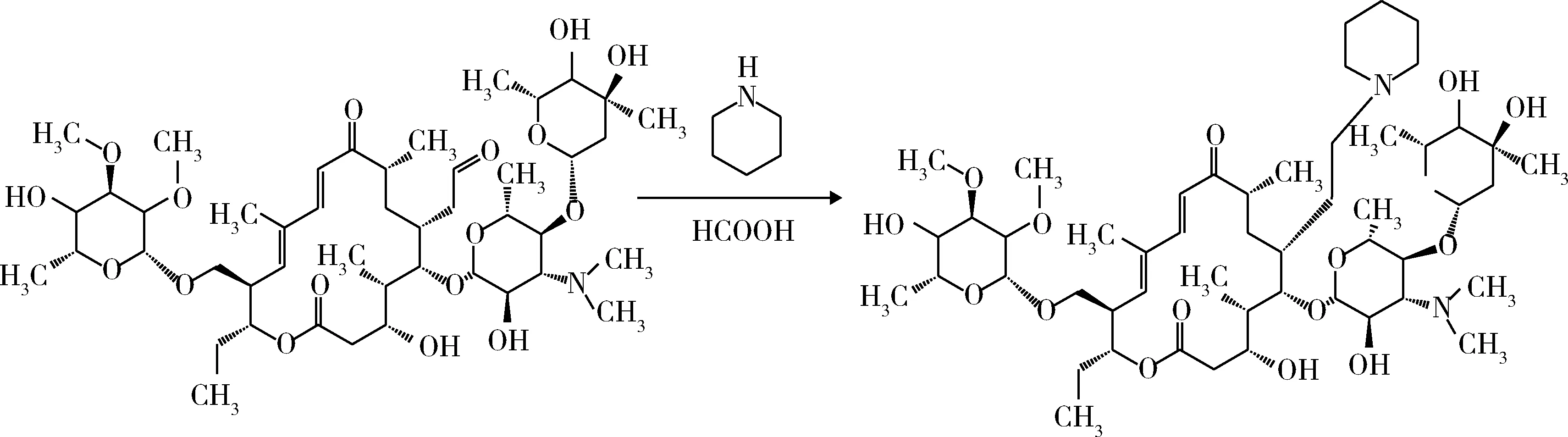
图4 23-哌啶基-泰乐菌素的合成Fig 4 Prepartion of 23-piperidin-tylosin
2.2 5-d-脱氧-阿洛糖的水解 向23-哌啶基-泰乐菌素碱的甲苯溶液内加入250 mL纯化水,使用42%的氢溴酸将水相pH调整至3~4之间,常温下搅拌萃取30 min,分去有机相。继续滴加42%氢溴酸,将pH调整至0.5,缓慢升温至57 ℃,保温反应5 h。反应结束后降至室温,加入200 mL二氯甲烷,用20%的NaOH溶液将pH调节至8.5,常温下萃取30分钟,分去水相,得到20-羟基-23-哌啶基-5-O-碳酶氨糖-泰乐内酯二氯甲烷溶液(图5)。
图5 20-羟基-23-哌啶基-5-O-碳酶氨糖-泰乐内酯的合成Fig 5 Prepartion of 5-O-Mycarose tylosin
2.3 TEMPO选择性氧化 在500 mL单口烧瓶中加入KBr20 g,再投入10 g的NaClO,加入100 mL纯化水,搅拌至全部溶解,用饱和NaHCO3将溶液pH调节至8.0,得到NaClO/KBr的缓冲溶液。将20-羟基-23-哌啶基-5-O-碳酶氨糖-泰乐内酯二氯甲烷溶液在氮气保护下降温至0 ℃,加入3.460 g(0.024 mol)2,2,6,6-四甲基哌啶氮氧化物,保温0 ℃并保持氮气保护下,充分搅拌至溶液为均匀的暗红色。缓慢滴加NaClO/KBr的缓冲溶液,直至溶液由暗红变为明亮的橙黄色。此时C-20为羟基被选择性氧化为羰基,得到20-羰基基-23-哌啶基-5-O-碳酶氨糖-泰乐内酯二氯甲烷溶液(图6)。

图6 20-羰基-23-哌啶基-5-O-碳酶氨糖-泰乐内酯的合成Fig 6 Prepartion of 20-carbonyl-5-O-Mycarose tylosin
2.4 C-20羰基取代 20-羰基基-23-哌啶基-5-O-碳酶氨糖-泰乐内酯二氯甲烷溶液氮气保护下升温至30 ℃,加入1.89 g(0.022 mol)哌啶,混合均匀后,升温至400 ℃。缓慢滴加1.11 g(0.024 mol)甲酸,滴加完毕后保温反应240 min。降温至室温,加入100 mL纯化水,用24%的氢溴酸调整pH至4.0,萃取30 min,分去有机相。再加入100 mL醋酸丁酯,用20%的NaOH调节pH至8.0,萃取30 min,分去水相, 将有机相蒸干,得到泰地罗新(图7)。
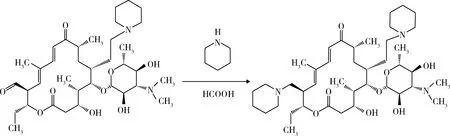
图7 泰地罗新的合成Fig 7 Prepartion of tildipirosin
3 结果与分析
研究的关键在于对C-20的选择性氧化,因此需要对本步骤的产物,即关键中间体20-羰基基-23-哌啶基-5-O-碳酶氨糖-泰乐内酯进行结构确认。同时通过液相色谱法,确定终产物泰地罗新的收率。
3.1 鉴别 为确认实验结果,研究对关键中间体20-羰基-23-哌啶基-5-O-碳酶氨糖-泰乐内酯进行结构确认,分别以液质联用与核磁共振确认了关键中间体结构,验证反应可行性,结果如表3、表4所示。

表3 液质联用分子量分布数据Tab 3 Liquid lc/molecular weight distribution data
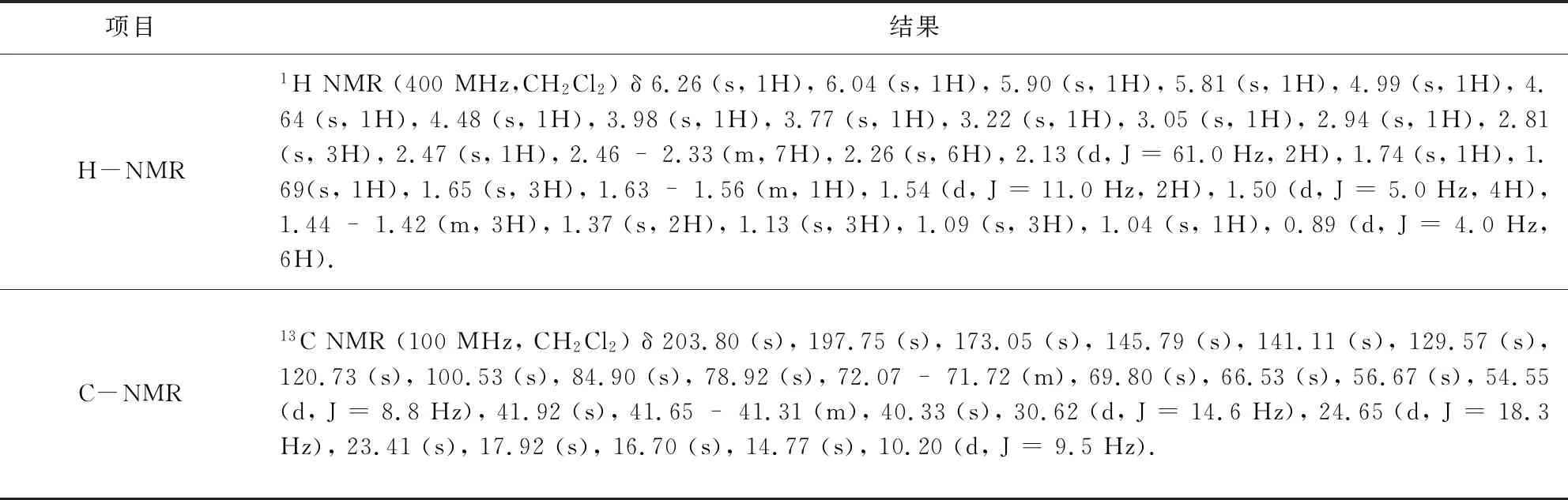
表4 核磁共振结果Tab 4 Nuclear magnetic resonance (NMR) results
3.2 反应得率 研究使用外标法测定了终产物泰地罗新的含量(图8),采用280 nm吸收波长,流动相采用乙腈∶甲醇∶0.05 mol/L磷酸二氢钾=3∶3∶4的混合溶液,固定相采用C18 3.5 μm色谱柱进行检测。标准品标称含量992 mg/g。

图8 泰地罗新HPLC图谱Fig 8 HPLC figure of tildipirosin
经检测,样品在14.770 min出峰,与标准品出峰时间一致,峰面积96.4%,外标法计算样品含量为971 mg/g,以此数据核算产物收率,反应总摩尔收率为90.11%。
4 讨论与结论
作为新一代十六元大环内酯类抗生素产品,泰地罗新的产业化生产越来越受到业内的关注。传统的合成路线受到原料成本高、反应条件苛刻等客观因素的影响,导致产品成本始终居高不下,使这一原本用于大规模养殖业的高效抗生素难以向市场大规模推广。
实验结果表明,采用TEMPO氧化路线得到的产物分子量、碳氢谱分析结果与20-羰基-23-哌啶基-5-O-碳酶氨糖-泰乐内酯完全相符,证明氧化法合成20-羰基基-23-哌啶基-5-O-碳酶氨糖-泰乐内酯的合成路线是可行有效的。新路线不使用昂贵的碘化剂,也规避了碘化反应苛刻的反应条件带来的副反应影响,降低了整体反应成本。
通过对新氧化得到的C23位羰基与哌啶进行Wallach反应,得到泰地罗新与标准品出峰时间一致,峰面积96.4%,外标法计算样品含量为971 mg/g,以此数据核算产物收率,反应总摩尔收率为90.11%。进一步证明了本研究所设计合成路线的可行性。
研究有效利用了2,2,6,6-四甲基哌啶氮氧化物在多羟基化合物的位阻效应,在进行C23羟基氧化反应时不影响其他碳位的羟基,已达到选择性反应的目的。区别于传统的碘代工艺路线,反应条件温和,成本低廉,更适宜工业化生产。
猜你喜欢
当代化工研究(2023年1期)2023-02-08
生物学杂志(2022年5期)2022-10-20
健康体检与管理(2022年2期)2022-04-15
陶瓷学报(2021年5期)2021-11-22
山东工业技术(2016年15期)2016-12-01
合成化学(2015年1期)2016-01-17
应用化工(2015年9期)2015-12-24
应用化工(2014年1期)2014-08-16
中国药理学通报(2014年2期)2014-05-09
郑州大学学报(理学版)(2014年4期)2014-03-01
