
Structural,Thermodynamical and Electronic Properties of All-Inorganic Lead Halide Perovskites
2021-06-04 03:49YawenLiGuangrenNaShulinLuoXinHeLijunZhang
物理化学学报 2021年4期
Yawen Li ,Guangren Na ,Shulin Luo ,Xin He ,2,*,Lijun Zhang ,*
1 State Key Laboratory of Integrated Optoelectronics,Key Laboratory of Automobile Materials of MOE,College of Materials Science and Engineering,Jilin University,Changchun 130012,China.
2 College of Physics,Jilin University,Changchun 130012,China.
Abstract:Organic-inorganic hybrid lead halide perovskites have emerged as the most promising materials in the field of optoelectronics due to their unique electronic and optical properties.However,the poor long-term material and device stabilities of these materials have limited their practical application.Compared to organic-inorganic hybrid perovskites,all-inorganic halide perovskites like CsPbX3 (X = Cl,Br,I)show enhanced thermal stability and the potential to resolve the issue of instability.Nevertheless,the structural and physical properties of allinorganic CsPbX3 halide perovskites with multiple structural polymorphs are still under debate.A recent research article on CsPbI3 reported the wrongly indexed the XRD pattern of γ-CsPbI3 as α-CsPbI3.Consequently,the band gap of γ-CsPbI3 (1.73 eV)was erroneously designated for α-CsPbI3.Therefore,there is a need for systematic research on the relationship between the structural features and electronic properties of CsPbX3.Here,we present a comprehensive theoretical study of the structural,thermodynamical and electronic properties of three polymorphic phases,α-,β-,and γ-CsPbX3.The space group of α-,β-,and γ-CsPbX3 are Pm3¯m,P4/mbm,and Pnma,respectively.First-principles calculations indicate that the phase transition from the hightemperature α-phase to the low-temperature β-phase and then to the γ phase is accompanied by an increase in the degree of PbX6 octahedral distortion.The zero-temperature energetic calculations reveal that the γ-phase is the most stable.This is consistent with the fact that experimentally,the γ-phase is stabilized at a relatively low temperature.Analysis of the electronic properties indicates that all the CsPbX3 perovskites exhibit a direct-gap nature and the band gap values increase from α to β,and then to the γ phase.From the analysis of the orbital hybridization near the band gap edges,the increase can be explained by the downshift of the valence band edges caused by the gradual weakening of the Pb-X chemical bond.Among all the phases,the strongest Pb-X interaction in the α-phase leads to the most dispersive band-edge states and thus the smallest carrier effective masses,which are beneficial for carrier transport.Additionally,the band gaps decreased by changing the halogen type from Cl to Br and I under the same phase.this is a consequence of the increased X np orbital energies from Cl 3p to Br 4p and then to I 3p that leads to a high valence band edge for CsPbI3 and results in the smallest band gap.Our results provide deep understanding on the relationship between the physical properties and structural features of all-inorganic lead halide perovskites.
Key Words:Inorganic halide perovskite; Optoelectronic property; First-principles calculation; Electronic structure
1 Introduction
Since the organic metal perovskite was first reported in 20091researches on perovskite-based solar cells (PSCs)have exploded due to their excellent characteristics such as tunable band gap,low cost,and outstanding optical absorption properties2–12.The record certainly power conversation efficiency (PCEs)of organic-inorganic based PSCs has reached 25.2%13.Despite the organic-inorganic hybrid halide perovskites have high efficiencies and excellent properties,the poor stability of these materials is still a critical challenge which hinders the commercial pace of PSCs14.Thus researches have been focused on the long-term stability of solar absorber materials15–23.Using more robust inorganic Cs+cations to replace the fragile organic group with forms the all-inorganic perovskites lead halide system (CsPbX3,X is halide)as which are much more thermally stable and often more stable to other factors24,25.Inorganic perovskites CsPbX3have attracted great attention in the fields of photovoltaics26–28.From the first report in 2015 to now,the PCE of CsPbX3-based PSCs has abruptly increased from 2.9%(CsPbI3)29and 5.8% (CsPbBr3)30to 17.1% (CsPbI3)31with much enhanced stability.From this progress,the all-inorganic halide perovskites CsPbX3are emerging as a new research area and will play a significant role in the photovoltaic field32.
It is generally known that the inorganic lead-halide perovskites CsPbX3(X = Cl,Br,I)undergo phase transitions at different temperatures.The temperature range for the phase transition is different with the change of halogen atom.CsPbX3perovskites exhibit different polymorphs:α-(cubic),β-(tetragonal),and γ-(orthorhombic)phases.For bulk CsPbCl3,the high temperature phase is cubic α perovskite structure (Pm¯m).As bulk CsPbCl3is cooled through 47 °C,it undergoes firstorder phase transition to the tetragonal β perovskite structure(P4/mbm)which accompany by the PbCl6octahedra rotation in the ab plane.Finally,upon cooling through 42 °C,the PbCl6octahedra tilt away from the c axis to generate the orthorhombic γ perovskite (Pnma)structure (second-order transition)33,34.The single crystal CsPbBr3undergoes the cubic to tetragonal structure at 130 °C and tetragonal to orthorhombic at 88 °C35,36.The phase transitions of CsPbI3from high temperature to room temperature at ambient conditions are going through cubic to tetragonal phase at 260 °C,tetragonal to orthorhombic phase at 175 °C and then orthorhombic phase transition to non-perovskite δ-phase after standing a few days later37.Concerning the cubic phase,the phase transitions are accompanied by lead-halide octahedral tilting and distortion38,which affect the stability,electronic and photophysical properties39.In addition,the halogen anions also affect the electronic properties of lead halide perovskite40.
Recent researches indicated that there are incorrect cognitions about the structure and band gap of CsPbI324,41,42.Most of the previous studies have translated the X-ray diffraction (XRD)pattern of γ-CsPbI3into α-CsPbI3then wrongly attributed the band gap 1.73 eV to α-CsPbI3.Up to now,systematic research of electronic properties on three polymorphic phases inorganic lead halide perovskites:α/β/γ-CsPbX3are rarely reported.For all these reasons,it is necessary to have a comprehensive study on the different phases of inorganic perovskite CsPbX3.In this work,the structural,thermodynamical and electronic properties were calculated by first-principles calculations.From our results,we notice that the phase transition from α to β and then to γ are accompany with PbX6octahedral tilting and distortion,which further influence the thermostability and band gaps.Here,we investigate the in-depth physical mechanisms for band gap evolutions with phase transitions and halogen variations.
2 Computational methods
All the first-principles calculations were carried out within density functional theory (DFT)by using plane-wave pseudopotentials method implemented in the Vienna Ab-initio Simulation Package43.The projected augmented wave (PAW)pseudopotentials44with 6s for Cs,6s and 6p for Pb,3s and 3p for Cl,4s and 4p for Br,and 5s and 5p for I valence electrons were employed to describe the electron-ion interactions.The generalized gradient approximation (GGA)formulated by Perdew,Burke,and Ernzerhof (PBE)45was used as the exchange-correlation functional.Structural optimizations were done with the kinetic energy cutoff of 300 eV and the k-point meshes of reciprocal spacing set to 2π × 0.00318·nm−1.Both atoms and cell volume were allowed to relax until the residual forces were smaller than 0.1 eV·nm−1.Standard DFT-GGA methods underestimate the band gaps due to the presence of artificial self-interaction and the absence of the derivative discontinuity in the exchange-correlation potential46.We calculated electronic structures using the Heyd-Scuseria-Ernzerhof (HSE)functional with the standard 25% nonlocal Fock exchange.We also considered the spin-orbit coupling(SOC)in our calculation due to the non-negligible effect on the heavy metals,especially Pb.
3 Results and discussion
3.1 Structural properties and thermodynamic stability
The α,β and γ phases of CsPbI3and CsPbBr3are found to be in the Pmm,P4/mbm and Pnma space groups,respectively.There is experimental research on CsPbCl347revealed that the second and first-order phase transitions from orthorhombic over tetragonal to cubic structure occur at 42 and 47 °C,respectively.The β-CsPbCl3is in space group tetragonal P4/mbm33and γ-CsPbCl3is in space group orthorhombic Pnma49,which are same to the space groups of β/γ-CsPbBr(I)3.Therefore,we constructed the β/γ-CsPbCl3by replacing I atoms to Cl atoms based on β/γ-CsPbI3structures,and then the structures are fully relaxed (Fig.1a–c).The optimized lattice constants and calculated band gaps are shown in Table 1.The corresponding experimental data are also list for reference24,36,40,48–54.The crystal structures,bond lengths and bond angles between Pb and X of α,β and γ-CsPbX3are shown in Fig.1.As seen in Fig.1d,α-phases have the smallest Pb-X bond length and γ-phases have the largest bond length.In addition,the Pb-X bond length changing with halogen in the following order:CsPbCl3> CsPbBr3> CsPbI3.The PbX6octahedral framework is the skeleton of CsPbX3perovskite.The deviation of the band angles in the PbX6octahedrons (X-Pb-X,θ1)from 90° is taken as a measure of octahedral distortion.The deviation of the bond angles between corner-sharing PbX6octahedrons (Pb-X-Pb,θ2)from 180° is taken as a measure of octahedral tilting and twisting.As seen in Fig.1e,for β-phase,zero deviation of θ1and nonzero deviation of θ2indicating octahedron tilting.For γ-phase,the nonzero deviation of both θ1and θ2indicating octahedral distortion and octahedral tilting.Overall,phase transition accompanied with lead-halide octahedral tilting and distortion.γ-phase has the most distortion,followed by β-phase.

Fig.1 (a–c)The geometry structures of α/β/γ-phase CsPbX3 (X = Cl,Br,I),(d)the bond lengths of Pb-X and (e)the bond angles in the PbX6 octahedrons (X-Pb-X,θ1)and between PbX6 octahedrons (Pb-X-Pb,θ2).

Table 1 The optimized structural parameters and calculated band gaps by difference functional.
The thermodynamic stabilities of all-inorganic perovskites CsPbX3were analyzed by evaluating the formation energies.The formation energies are shown in Fig.2.Based on our calculations,on the one hand,γ-phase has the lowest formation energy,this is consistent with the experimental observation that γ-CsPbCl3is the phase stabilized at the lowest temperature range.On the other hand,CsPbCl3has the lowest formation energy compared to CsPbBr3and CsPbI3,which make it easier to synthesis in experiment.

Fig.2 Calculated formation energies of α/β/γ-CsPbX3.
It should be emphasized that the free energies were calculated at zero temperature.When temperature effect is included,the free energy will include the contributions such as thermal expansion,lattice vibrations,entropy effect,etc.First-principle study55found that the lattice vibration is the major contributing factor for stabilizing the cubic phase at room and high temperatures.We can expect that if the temperature effect is included,the energy order would be changed and the α/β-phase would be stabilized.
3.2 Electronic structures and band gaps
Fig.3 shows the calculated electronic structures by using standard HSE functional with considering SOC effects (HSE +SOC).It worth noting that the normal first-principles DFT calculation usually underestimates the band gap because of the self-interaction error.Generally,this issue could be remedied by using the HSE functional.For the Pb-containing perovskites,however,the band gaps calculated by HSE + SOC still do not agree well with the experimental values56.In this work,the band gap evolution of α/β/γ-CsPbI3calculated by HSE + SOC are consistent with the experimental trend (see Table 1).We therefore adopted the HSE + SOC results to analyzed band gap evolutions with phase transitions and halogen variations.All these nine structures are direct band gap semiconductors.The band edges of three α-phase structures are locating at (0.5,0.5,0.5)k-point,and three β- and γ-phase structures are locating at(0,0,0.5)and (0,0,0)k-points.The orbital compositions of the band edge states are shown.From the projected band edges,we can see that the valence-band maximums (VBMs)are mainly composed of the antibonding hybrid states between Pb 6s and X np (X = Cl,Br,and I)orbitals.The conduction band minimums(CBMs)are contributed by the antibonding states between Pb 6p and X np orbitals,with the major contribution from Pb 6p.Cs+has no significant contribution around the band edge.This is further reproduced by the project density of states (PDOS)shown in Fig.S1 (Supporting Information),we can see that the CBM mainly contributed by the Pb 6p,while the ratio of I 5p state is rather limited.It should be noticed that the band gaps of these structures present a monotonic change with phase transitions and halogen variations.Firstly,the band gaps increase monotonically with the phase transition from α- to β- and to γphase.Secondly,the band gaps decreased by changing the halogen type from Cl to Br and to I.

Fig.3 The HSE + SOC functional calculated band structures of (a–c)α/β/γ-CsPbCl3,(d–f)α/β/γ-CsPbBr3 and (g–i)α/β/γ-CsPbI3.The dominant orbital characters of the bands are shown in color.Red,green and blue lines represent Pb s,Pb p and X p states,respectively.
In order to have a deeper understanding of the physical mechanisms for band gap evolution with phase transitions,we further investigated the orbital coupling between the Pb cation and halide ion around the band edges in Fig.4a.For the example of CsPbI3,the PbI6octahedral structures and partial charge density at the VBM of α,β and γ-CsPbI3were presented in Fig.4b–d.As we can see,from α-phase (0.319 nm of Pb-I bond length)to β-phase (0.322 nm)and then to γ-phase (0.325 nm).The bond length of Pb-I increases gradually,which lead to weakening of Pb-I interaction.As a result,the antibonding states of Pb 6s–I 5p (VBM)are suppressed54.Because the CBM is mainly contributed by the Pb 6p states,the influence of Pb-I bonding strength on the CBM position is relatively small.The band gap increment can be attributed to weakening of the Pb-I interaction with increasing of the bond length and PbI6octahedral distortion.

Fig.4 (a)The schematic of orbital hybridization between Pb cation and halide ion near the band edge states in CsPbI3.(b–d)The bond length diagram and charge density at valence band maximum (VBM)of Pb-I in cubic,tetragonal and orthorhombic CsPbI3,the isosurface value is set at 1 e·nm−3.
In addition,the band gap changing with halogen in the following order:CsPbCl3> CsPbBr3> CsPbI3.CsPbI3has the smallest band gap value in the CsPbX3with the same phase structure.On the one hand,this can be attributed to the relativity smallest electronegativity difference between Pb (2.33)and I(2.66)makes Pb-I bond more covalent in comparison with Pb-Br (Br:2.96)and Pb-Cl (Cl:3.16).The strong interaction pushes up the antibonding states of Pb 6s and I 5p (VBM).On the other hand,The X np orbital energies are decreased from I 5p to Br 4p and then to Cl 3p,which leads to a high VBM for CsPbI3and results in the smallest band gap value of CsPbI3.
It is worth mentioning that recent research for the CsPbI324reported the wrongly indexed the XRD pattern of γ-CsPbI3into α-CsPbI3.As a result,the band gap of γ-CsPbI3(1.73 eV)is erroneously referred to α-CsPbI3.As for the CsPbCl3and CsPbBr3,the experimental band gap values of cubic phases are larger than that of the orthorhombic phases.However,our calculated results and the bonding analysis demonstrate that the band gaps of orthorhombic phases should be larger than that of cubic phases.Therefore,the electronic properties of CsPbBr3and CsPbCl3should be further studied and established through experimentation.
3.3 Carrier effective masses
Carrier mobility is crucial for perovskite solar cell applications.As is well known,the effective masses of electron and hole are closely related to carrier mobility57,58.The calculated transport effective masses for electron me*and mh*are shown in Fig.5.As seen,all these materials have low electron and hole effective mass in the range of 0.05m0–0.15m0,which are consistent with other theoretical studies (eg.mh*= 0.073m0and me*= 0.064 m0for α-CsPbI3)59.These values are even lower than those of inorganic-organic halide perovskite CH3NH3PbX3(mh*= 0.23m0and me*= 0.29m0)60.It is beneficial for carrier transport.Moreover,α-CsPbX3have the smallest effective mass than other phases.Take CsPbI3as an example,α-CsPbI3has the smallest Pb-I bond length.The strong Pb-I orbital interaction leads to a more dispersive band-edge state and results in smaller effective masses for α-phase.The effective masses along three directions of electron and hole also calculated and shown in Table S1 (Supporting Information).
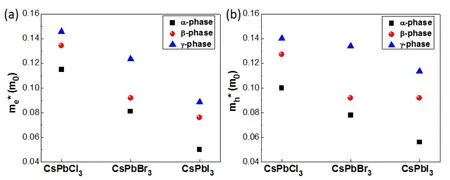
Fig.5 The HSE + SOC functional effective masses of electron (a)and holes (b)of three phases inorganic perovskites.
4 Conclusions
In this work,we present a comprehensive study for the structural,thermodynamical and electronic properties of all inorganic lead halide perovskites (α-,β- and γ-CsPbX3(X = Cl,Br,I))by performing first-principles calculations.Phase transitions are accompanied by PbX6octahedral tilting and distortion,which influence the thermal stability and electronic properties.γ-phase with the lowest formation energy is the most stable structure,consistent with the fact that it is stabilized at relatively low temperature in experiment.All the CsPbX3perovskites are direct-gap semiconductors and the band gap increasing with the phase transitions from α- to β- and then to γphase.This is driven by the weaker Pb-X interaction due to the octahedral tilting and distortion,thus reducing the valence band maximum (VBM,consist of antibonding states of Pb 6s and X np orbitals).In addition,the band gap decreases from Cl to I in CsPbX3under the same structure.This is mainly due to the relative positions of the halogen np orbital energies being gradually increased from Cl to I and thus increased the VBM.All these compounds have favorable transport effective mass values in the range of 0.05m0–0.15m0.Our work provides understanding on the relationship between physical properties and structural features for all inorganic lead halide perovskites.
Supporting Information:available free of charge via the internet at http://www.whxb.pku.edu.cn.

- 物理化学学报的其它文章
- SCN-doped CsPbI3 for Improving Stability and Photodetection Performance of Colloidal Quantum Dots
- 两步互扩散法制备高性能CsPbCl3薄膜紫外光电探测器
- 电泳法制备的致密氧化锡薄膜及其在高稳定性钙钛矿太阳能电池中的应用
- Highly Moisture Resistant 5-Aminovaleric Acid Crosslinked CH3NH3PbBr3 Perovskite Film with ALD-Al2O3 Protection
- 基于易升华添加剂辅助合成纯相富铯CH(NH2)2)xCs1−xPbI3钙钛矿
- 锡基钙钛矿太阳能电池研究进展