
气相色谱-质谱法测定植物蛋白饮料中植物甾醇和胆固醇
2021-06-04 14:03周蕾
中国酿造 2021年5期
周 蕾
(北京市朝阳区食品药品安全监控中心,北京 100123)
植物蛋白饮料是以一种或多种含有一定蛋白质的植物果实、种子或种仁等为原料,经加工或发酵制成的一类饮品[1],其保留了原料中所富含的营养素、生物活性物质和植物化学物质,不仅可部分替代动物奶,以满足奶制品过敏者及乳糖不耐受者对蛋白质的需求,还可作为植物源蛋白饮食中的组成部分来减少疾病和肥胖的发生[2-4],因此近年来备受消费者的青睐。目前,对植物蛋白饮料的技术研发多集中于生产工艺[5]、产品稳定性[6]、理化指标[2-3]和掺假鉴定方法[7]等,而对其生物活性成分的研究较少,尤其缺乏关于植物蛋白饮料中植物甾醇含量的测定和特征分析。
植物甾醇是一类以环戊烷多氢菲为骨架、结构类似于胆固醇的植物活性物质,主要以游离、酯化或糖苷等形式天然存在于植物源食物中,尤以植物油、坚果和豆类中含量最高,其植物甾醇总量分别可达1 055 mg/100 g(米糠油)、158 mg/100 g(腰果)、135 mg/100 g(豌豆)[8-9]。根据分子结构中双键数量、双键的位置及C24上烷基的差异,可将植物甾醇细分为不同的种类,如今已鉴定出超过250种植物甾醇及相关化合物,其中最为常见、含量比较高的为β-谷甾醇、菜油甾醇和豆甾醇等[10-13]。欧洲食品安全局(European Food Safety Authority,EFSA)和美国食品药品监督管理局(food and drug administration,FDA)研究表明,与安慰剂相比,人体每天摄入1.5~3.0 g植物甾醇和甾烷醇(以游离甾醇计),血液中的低密度脂蛋白(low-density lipoprotein,LDL)胆固醇水平可降低7.0%~12.5%[14-15]。因此,已有多个国家批准植物甾醇及植物甾醇酯在多类食品中添加使用,我国也于2010年将植物甾醇及植物甾醇酯纳入了新食品原料名录[16]。
目前用精密仪器对植物甾醇和胆固醇进行准确定量测定的方法有气相色谱法[12]、液相色谱法[17-19]、气相色谱-质谱法(gas chromatography-mass spectrometry,GC-MS)[20-22]、气相色谱-串联质谱法[23-24]、液相色谱离子阱质谱法[25]、超高压液相色谱质谱法[26]、液相色谱串联质谱法[27-28]、近红外法[29]等。液相色谱质谱/串联质谱法具有无需衍生、灵敏度高的优势,但仪器普及率较低、操作复杂、耗费较大[25-26,30]。近红外法无需样品前处理,为食用油中植物甾醇的测定提供了一种在线、无损、快速的检测方法,但低含量样品中豆甾醇的测定结果准确度欠佳[29]。气相色谱质谱法选择性好、灵敏度高、抗干扰能力强,日益成为测定食品中植物甾醇含量最常用的方法。
本研究拟采用气相色谱质谱技术,以5α-胆甾烷为内标,建立同时测定植物蛋白饮料中β-谷甾醇、豆甾醇、菜籽甾醇、菜油甾醇、豆甾烷醇5种植物甾醇及胆固醇(动物源奶标记物)含量的方法,可评估植物蛋白饮料中植物甾醇含量的同时,对该产品中动物源奶配料情况进行监测。旨在为植物蛋白饮料产品开发、质量控制、功能评价等研究提供一定的技术基础。
1 材料与方法
1.1 材料与试剂
5α-胆甾烷、豆甾醇、β-谷甾醇、菜油甾醇(纯度均≥97.0%):上海安谱实验科技股份有限公司;胆固醇、豆甾烷醇(纯度均≥98.0%):美国Sigma-Aldrich公司;菜籽甾醇(纯度≥98.0%):加拿大Toronto Research Chemicals公司;正己烷(色谱纯):赛默飞世尔科技(中国)有限公司;N,O-双(三甲基硅)三氟乙酰胺-三甲基氯硅烷(bis(trimethylsilyl)trifluoroacetamide∶trimethylchlorosilane,BSTFA∶TMCS=99∶1)衍生化试剂:美国REGIS Technology公司。无水乙醚(优级纯):天津市科密欧化学试剂有限公司;石油醚(30~60 ℃)、氢氧化钾、无水乙醇、无水硫酸钠(均为分析纯):国药集团化学试剂有限公司。
植物蛋白饮料样品,样品包括2个豆奶(1#、2#)、2个椰子汁(3#、4#)、1个核桃乳(5#)和1个杏仁露(6#):市售。
1.2 仪器与设备
Clarus 600 Gas Chromatograph-Clarus 600T Mass Spectrometer气相色谱质谱联用仪(配备Clarus autosampler自动进样器):美国PerkinElmer公司;Milli-Q Reference超纯水系统:美国Millipore公司;Elmasonic P 300H数控超声波清洗器:Elma-Hans Schmidbauer GmbH&Co.KG;Multifuge X1R高速冷冻离心机:德国ThermoFisher公司;OA-SYS Heating System N-EVAP 112氮气吹干仪:美国Organomation Associ ates,Jnc公司;TLE204电子分析天平:梅特勒-托利多仪器(上海)有限公司;DH-101-2BS电热恒温鼓风干燥箱:天津市中环实验电驴有限公司。
1.3 方法
1.3.1 标准溶液配制
分别称取10 mg(精确至0.01 mg)5α-胆甾烷、胆固醇和植物甾醇标准物质至10 mL容量瓶中,用正己烷溶解并定容,混匀后配制成质量浓度为1.0 mg/mL的内标标准使用液和甾醇标准储备液。
1.3.2 样品处理方法
按照参考文献[21]中前处理方法并略有改动:称取0.5 g(精确至0.000 1 g)植物蛋白饮料样品于10 mL离心管中,依次加入5 μL 5α-胆甾烷内标使用液,3 mL 4 mol/L氢氧化钾-乙醇溶液,置于85 ℃恒温箱中皂化1 h后冷却至室温。然后加入2 mL去离子水和3 mL正己烷,涡旋2 min后4 000 r/min离心4 min,转移上层有机相至另一离心管中,重复提取2次,合并有机相,用去离子水洗至中性,经无水硫酸钠脱水后,氮吹浓缩至约1 mL,转移至进样小瓶中氮吹至近干,加入20 μL BSTFA:TMCS衍生化试剂,于室温(25 ℃)条件下衍生40 min,用正己烷定容至1 mL,上机分析。
1.3.3 仪器条件
色谱条件:Agilent J&W DB-5 MS毛细管色谱柱(30 m×0.25 mm×0.25 μm);进样口温度:250 ℃;载气:高纯氦气(纯度≥99.999%),初始压力152.3 kPa,载气流速16.2 mL/min,柱流速1.20 mL/min;分流进样,分流比10∶1,进样量1 μL;升温程序:初始温度250 ℃,以20 ℃/min升至300 ℃,再以5 ℃/min升至320 ℃,保持3 min。
质谱条件:电子轰击(electron impact ion source,EI)离子源,能量70 eV;离子源温度230 ℃;质谱接口温度260 ℃;溶剂延迟时间3 min;扫描模式:全离子扫描(Full Scan,扫描范围m/z 50.0~500.0)、选择离子扫描(selected ions monitoring,SIM)。
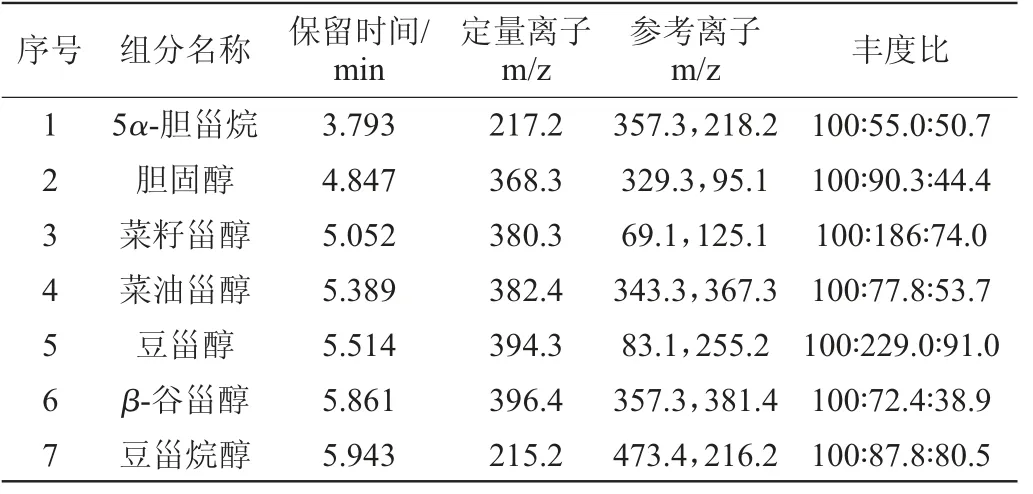
表1 5种甾醇和胆固醇及5α-胆甾烷气相色谱-质谱参数Table 1 Parameters of GC-MS of five phytosterols,cholesterol and 5α-cholestane
1.3.4 数据处理
采用TurboMass工作站(配备美国国家标准与技术研究院(national institute of standards and technology,NIST.14谱库)采集谱图及处理峰数据。实验数据采用Excel 2010软件进行分析和绘图。
2 结果与分析
2.1 仪器条件优化
针对气相色谱的分流进样、进样口温度、升温程序等参数进行对比分析,进样口温度250 ℃、分流比10∶1、柱流速1.2 mL/min、升温速率5 ℃/min条件下5种植物甾醇和胆固醇及5α-胆甾烷(内标)定量离子色谱图见图1。结果发现不分流进样时胆固醇和菜籽甾醇、菜油甾醇和豆甾醇、β-谷甾醇和豆甾烷醇的定量离子峰无法有效分离,且峰型较差。分流进样时峰型好、分离度高,随着分流比的增大,目标峰响应值变小,因此选择分流比为10∶1的分流进样。对比不同进样口温度,250 ℃时组分峰面积略大于220 ℃时组分峰面积,但随着温度从250 ℃提高至300 ℃时,除β-谷甾醇和豆甾烷醇峰面积先增后减外,其他组分均略微逐渐减小,因此选定进样口温度为250 ℃。随着柱流速的增加及升温程序中升温速率的提高,各组分峰的出峰时间逐渐提前,当以2 ℃/min升温至320 ℃时,目标峰能够与硅烷杂质峰完全分开,而以5 ℃/min升温时,两者虽完全重合,但质谱具有极高的选择性,经提取特征离子后,并不影响定性及定量,最终选择柱流速为1.2 mL/min、升温速率为5 ℃/min,此时单样品分析时间仅需9.5 min,大大提高了检测效率。

图1 5种甾醇和胆固醇及5α-胆甾烷内标定量离子色谱图Fig.1 Quantitative ion chromatograms of five phytosterols,cholesterol and 5α-cholestane
2.2 皂化及提取前处理
取样量为0.5 g,并参照文献[18]提高氢氧化钾-乙醇溶液浓度至4 mol/L,结果显示皂化效果良好。对比正己烷和乙醚∶石油醚(1∶1)两种提取溶剂,当皂化完成,加2 mL去离子水后用乙醚∶石油醚提取时,出现乙醇共提现象,虽然在随后用去离子水洗涤提取溶剂时随水相与乙醚∶石油醚层分离,但会造成组分流失,而正己烷可与乙醇+水相完全分层,因此选择正己烷作为提取溶剂。
2.3 甾醇组分衍生化方法
甾醇类组分沸点高、挥发性差、在高温下容易失水或者分解,在质谱中离子化能力差,因此信号相对较弱或不产生信号(见图2A),导致分析灵敏度、重现性不理想。为解决这些问题,通常对其进行衍生化处理,其中硅烷化试剂最为常用:BSTFA上甲基硅烷基和甾醇上的羟基发生氢交换,生成三甲基硅烷化甾醇,其极性降低,热稳定性更好,离子化能力增强,检测灵敏度提高[26,30](见图2B)。

图2 5种甾醇和胆固醇及5α-胆甾烷内标混合标准溶液未衍生化(A)和衍生化(B)全扫描色谱图Fig.2 Full scan gas chromatograms of five phytosterols,cholesterol and mixed standards of 5α-cholestane after underivatization(A) and derivatization (B)
衍生化效率与试剂加入量、反应温度和衍生时间相关,以10 mg/L甾醇标准溶液为对象,以进样小瓶为反应容器,经氮气吹干后分别加入5 μL、10 μL、20 μL、30 μL、100 μL试剂衍生,结果发现,衍生组分峰面积并无明显区别,为保证衍生充分,选定20 μL的衍生试剂加入量。为提高衍生化效率,通常采用保温加速衍生化反应[13,20-21,24],以及通过静置过夜来保证甾醇组分完全衍生化[20],对比室温(25 ℃)、60 ℃、80 ℃、105 ℃保温条件下衍生化效果,以保温时间为横坐标,以5α-胆甾烷内标校正后的各甾醇组分峰面积与最大值的比值为纵坐标作二维坐标图,如图3所示。

图3 不同衍生温度条件下甾醇衍生速率图Fig.3 Derivatization rate of sterol at different derivation temperature
由图3可知,随着衍生时间延长,衍生化率逐渐提高至稳定,且保温的温度越高,其衍生速率越高,越快达到稳定的最大值。但相比于常温,经内标校正后的各保温条件下衍生组分峰面积略小,可能是因为高温促进甾醇衍生化物挥发所致。而在TMCS的催化下,BSTFA即可在室温常压下对目标组分进行快速的衍生化反应,同时为减少保温的操作步骤,本试验选择室温(25 ℃)条件下衍生不少于40 min的衍生条件。
2.4 线性范围、检出限和定量限
分别取5 μL、10 μL、20 μL、50 μL、100 μL、200 μL、400 μL、800 μL各甾醇标准储备液至10 mL容量瓶中,各加入50 μL内标使用液,用正己烷定容。准确吸取1 mL各浓度标准溶液至进样小瓶中,经氮气吹干溶剂后,加入20 μL衍生化试剂,密封并静置40 min后用正己烷定容至1 mL,得到质量浓度为0.5~80.0 mg/L的标准工作溶液。经上机分析,以甾醇组分与内标的质量浓度比值为横坐标、相应的峰面积比值为纵坐标绘制标准曲线(见表2)。由表2可知,5种植物甾醇和胆固醇组分在各自线性范围内线性关系良好(R2≥0.999 9)。逐级配制低浓度甾醇标准溶液,经衍生后上机分析,分别以3倍信噪比(S/N)和10倍S/N计算方法检出限和定量限,结果表明,5种植物甾醇和胆固醇组分方法检出限为0.032~0.058 mg/kg,定量限为0.104~0.190 mg/kg。
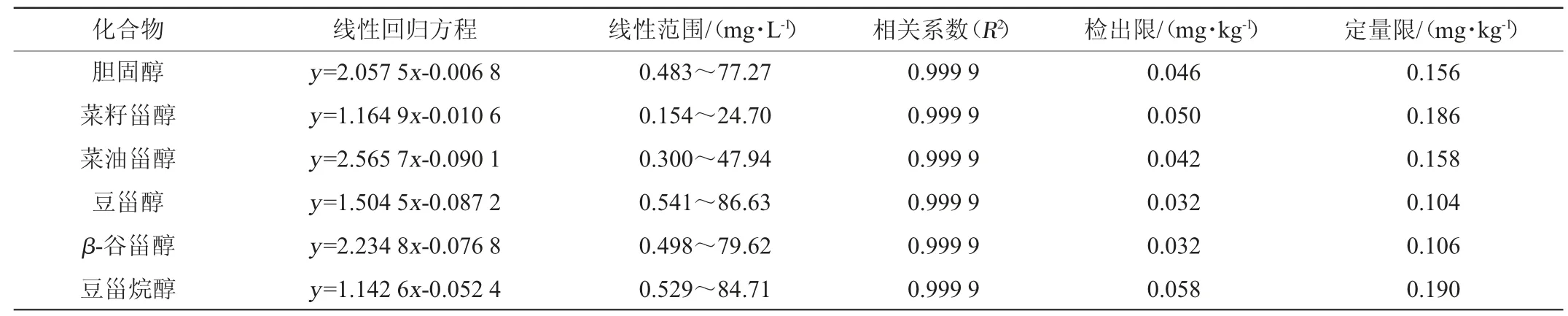
表2 5种甾醇和胆固醇的标准曲线回归方程、线性范围、线性方程、相关系数、检出限和定量限Table 2 Linear ranges,linear equations,correlation coefficients (R2),LODs and LOQs of the five phytosterols and cholesterol
2.5 加标回收率与精密度
采用低、中、高(以线性范围最低点浓度折算到样品中浓度的1倍、4倍、20倍)3水平加标方式,分别选用经确认的植物甾醇空白样品(牛奶)和胆固醇空白样品(纯大豆豆奶)作为加标对象,对植物甾醇和胆固醇进行加标回收试验,每一加标水平平行测定6次,计算平均回收率结果见表3。
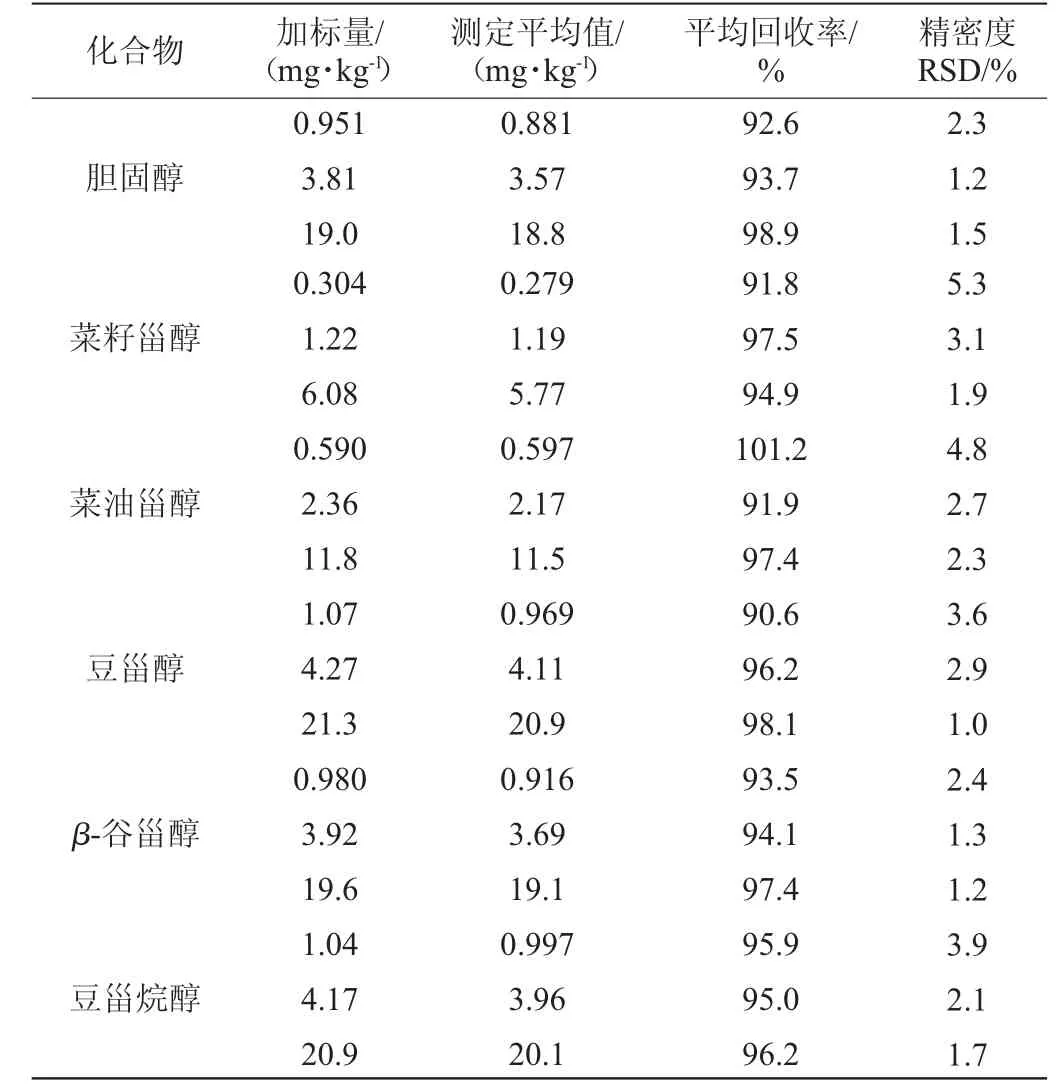
表3 胆固醇和植物甾醇的加标回收率和精密度试验结果Table 3 Results of adding standard recovery and precision of cholesterol and phytosterols
由表3可知,在0.304~21.3 mg/kg质量浓度范围内,5种植物甾醇和胆固醇的加标回收率为90.6%~101.2%,精密度相对标准偏差(relative standard deviation,RSD)为1.0%~5.3%。表明该方法的准确度和精密度良好,可以满足植物蛋白饮料样品中5种植物甾醇和胆固醇的同时测定。
2.6 样品测定与分析
由表4可知,6种样品均含有β-谷甾醇、菜油甾醇和豆甾醇,且均以β-谷甾醇为主,杏仁露和核桃露中还含有一定的豆甾烷醇,所测6种样品中胆固醇和菜籽甾醇含量均未检出。豆奶样品的植物甾醇总量相对较高,均大于60 mg/kg,而核桃露和杏仁露含量较低,这取决于原料自身植物甾醇的含量及原料的使用量。总体而言,植物蛋白饮料中植物甾醇含量虽远低于植物油中含量但其不含胆固醇,就日常饮用量计算得植物甾醇摄入量也远低于膳食推荐日摄入量(160~400 mg),但相比于其他饮料,植物蛋白饮料仍不失为日常饮食中植物甾醇的有效来源。
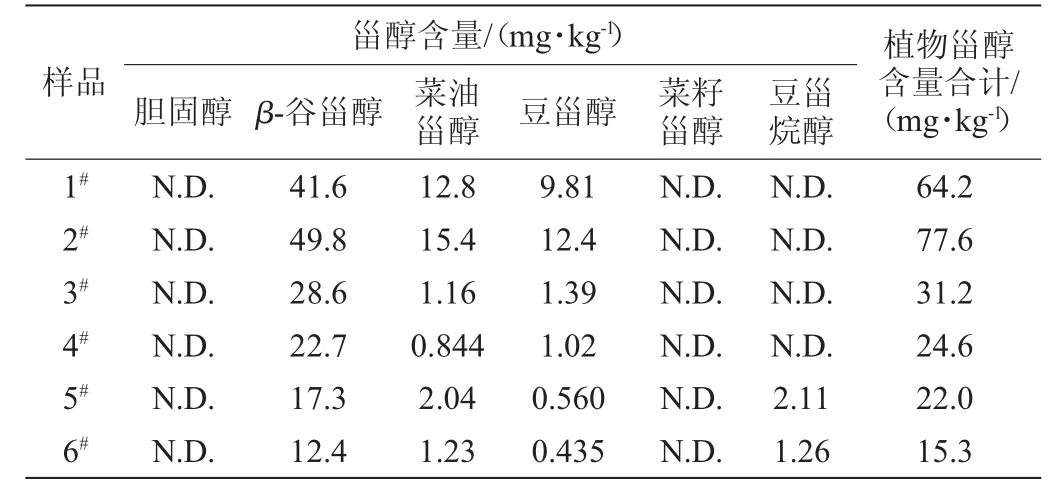
表4 实际样品中植物甾醇和胆固醇含量Table 4 Contents of phytosterols and cholesterol in samples
3 结论
本研究建立了同时测定植物蛋白饮料中胆固醇和5种常见植物甾醇的GC-MS方法,样品经皂化后对甾醇类化合物进行提取和衍生化,经质谱SIM模式内标法定量测定,5种植物甾醇和胆固醇的方法检出限为0.032~0.058 mg/kg,定量限为0.104~0.190mg/kg,标准曲线线性良好(R2≥0.9999),在低、中、高添加浓度范围内,加标回收率为90.6%~101.2%,精密度试验结果相对标准偏差为1.0%~5.3%。采用该方法对市售植物蛋白饮料进行分析,不同品种样品间植物甾醇含量存在明显差异。该方法操作简单、稳定性好、灵敏度高,适用于植物蛋白饮料中植物甾醇和胆固醇的测定。
猜你喜欢
食品安全导刊(2022年32期)2022-12-07
石油炼制与化工(2022年2期)2022-02-15
口腔护理用品工业(2021年4期)2021-11-02
化工管理(2020年26期)2020-10-09
山东化工(2019年2期)2019-02-21
中成药(2018年6期)2018-07-11
中国粮油学报(2017年5期)2017-07-19
电子技术与软件工程(2016年24期)2017-02-23
中国粮油学报(2016年5期)2016-01-23
长江大学学报(自科版)(2014年27期)2014-02-27
