
水泥矿物体系诱导期的水化进程及机理的研究进展
2021-08-10 01:38胡匡艺储静远艾晓晶盖琳琳盛嘉诚
硅酸盐通报 2021年7期
官 敏,胡匡艺,于 涛,储静远,艾晓晶,常 郑,盖琳琳,盛嘉诚
(中国建材国际工程集团有限公司,上海 200063)
0 引 言
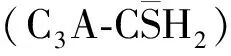
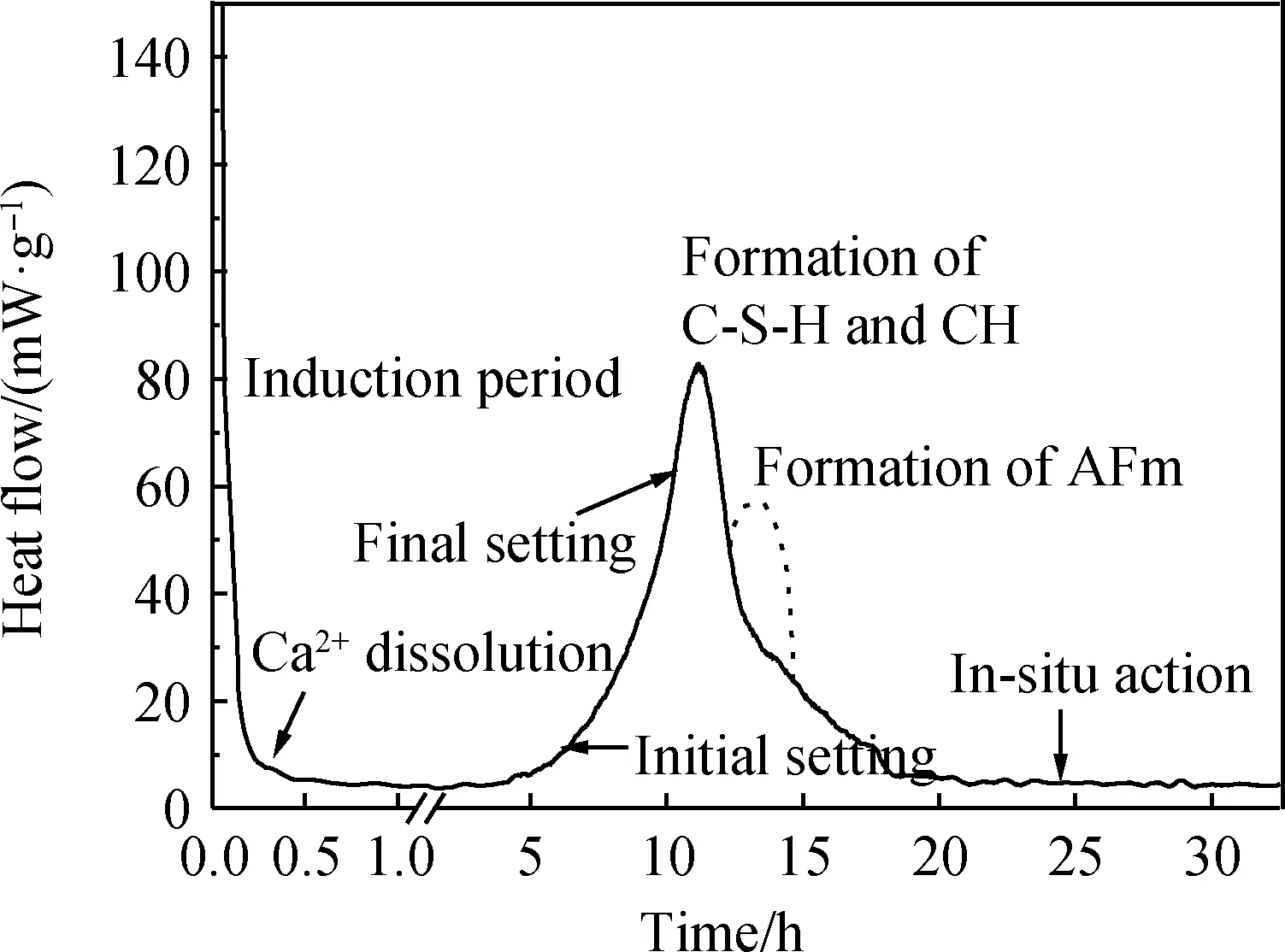
图1 水泥水化进程的划分[3]Fig.1 Hydration process division of Portland cement[3]
1 水化作用的热力学原理
水泥水化作用的基础是溶解-沉淀过程,未水化相与水化产物之间如果没有溶液中的离子扩散是不能发生转化的。水化进程中的水化产物必须具有比未水化相更低的溶解度。在氧化钙-氧化铝-水(CaO-Al2O3-H2O)三元体系中,溶解度最低的是3CaO·Al2O3·6H2O(C3AH6)。但是C3AH6的形成慢于溶解度更高的2CaO·Al2O3·8H2O(C2AH8) 以及4CaO·Al2O3·13H2O(C4AH13),因此后者首先生成,提高温度则可以促进向C3AH6的转变[4],这表明结合热力学与动力学或许可以预测产物的生成。
但不同条件下物相溶解度的差异对研究多相系统又是不足的,例如对硅酸三钙-硅酸二钙(C3S-C2S)二元体系的研究[5]。C3S与C2S的溶解度均高于水化硅酸钙(C-S-H),但是在均匀混合的C3S-C2S二元体系中,只要C3S正在水化,C2S便无法溶解。这是因为C3S水化时溶液中的离子浓度高于C2S的溶解度。所以,了解物相的水化速率是必要的。
水化作用是一个复杂过程,至少涉及到两种固相(一种未水化相(反应相)和一种水化产物相(生成相))和一种液相。未水化相的溶解存在溶解速率Rd,水化产物的沉淀存在沉淀速率Rp。水化是一种界面反应,因此水化速率还与固相与液相的接触面积S有关。从热力学角度出发,偏离平衡越远,吉布斯自由能的绝对值ΔG越大,Rd和Rp越高。此外,溶液中的一些离子可能对表面反应具有特定的影响。在各向同性材料的情况下,t时刻的溶解或沉淀速率可写为[6]:
R(t)=rif(ΔG(t),[ions], …)S(t)
(1)
式中:R(t)为t时刻的溶解或沉淀速率,mol·s-1;rif为溶解或沉淀的界面速率,mol·m-2·s-1;[ions]为任意离子浓度,mol·L-1;S(t)为溶解或生成的表面积,m2。
rif与S均随时间变化。考虑到溶解时固体表面积的减少以及沉淀时固体表面积的增加,在水化进程中溶解界面速率和沉淀界面速率也是不同的,但在大部分时间里,溶解速率近似等于沉淀速率,即:
Rp≅Rd
(2)
由于在水化开始时溶解的表面积Sp为0,而在水化结束时生成的表面积Sd为0,为了满足式(2),反应相溶解的界面速率与生成相沉淀的界面速率的比值(rifd/rifp)必须持续增大。水化开始时,溶液中离子浓度近似于反应相的溶解度,而在水化结束前,溶液中的离子浓度接近生成相的溶解度。该过程被Barret[5]命名为水化作用的动力学过程。
水化作用的开始阶段很特别,除晶核的形成外,生成相还不存在,此时仅存在反应相单独溶解使足够的离子进入液相诱导新相(生成相)的产生。实际上,如果生成溶解度较低的新相使体系的自由能降低,那么在水化产物与低自由能的溶液之间会形成新的界面,形成界面所需的自由能来自溶液对于生成相的过饱和。根据经典形核理论,形成可以生长的稳定晶核所需的诱导时间tip取决于溶液过饱和度β(离子活度积Iap与平衡溶度积Ksp的比值) 以及晶体与溶液间的界面能γ[6],如式(3)所示:
(3)
式中:f为形核系数;Ω为摩尔体积;κ为玻尔兹曼常数;T为形核温度;K0为动力学常数。
溶液过饱和度越高,形核所需的诱导时间越短。如果反应相的溶解速率较高,并且反应相的溶解度高于生成相的溶解度,则可以更快达到生成相所需的过饱和度。C-S-H在C3S水化进程中形核的时间极短,几乎无法测量[7]。相反,C2S在氢氧化钙(CH)溶液中发生水化作用时,诱导期长达数十分钟,因此可以观察C-S-H的形核过程[8-9]。在铝酸一钙(CA)水化进程中,CaO·Al2O3·10H2O(CAH10)形核所需的时间可能更长[10-11]。

这些理论使理解和预测愈加复杂的胶凝材料体系随时间的水化进程成为可能。综上所述,大多数水泥水化进程可以满足下式[6]:
R(t)=rifd(t)Sd(t)=rifp(t)Sp(t)
(4)
式中:rifd(t)为t时刻未水化相溶解的界面速率;Sd(t)为t时刻未水化相溶解的表面积;rifp(t)为t时刻水化产物相沉淀的界面速率;Sp(t)为t时刻水化产物相生成的表面积。
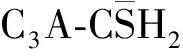
2 C3S体系的溶解
作为水泥中占比最大的组分,C3S的水化受到了极大的关注。C3S体系在水化进程中重要的特征之一是在加入水之后C3S反应速率迅速降低,导致水化过程减缓,一段时间后反应速率再次提升。诱导期具有重要的实际意义,可以提供在终凝之前开始运输和浇筑混凝土的时间。然而,诱导期在实际放热曲线中通常表现为最小值,如图2 C3S体系的水化放热曲线所示,并非图1中所示的明显平坦阶段。多年来,C3S水化的诱导期一直是学界关注的问题,并且许多学者提出了不同假说来解释其成因。其中介稳层假说和缓慢溶解假说受到广泛认同,这两种假说在大部分研究中具有较强的合理性。
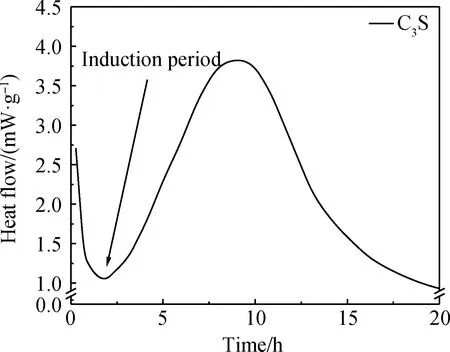
图2 C3S体系的水化放热曲线[11]Fig.2 Heat flow curve of C3S system hydration[11]
2.1 介稳层假说(metastable barrier hypothesis)
Stein[13]及Jennings[14]等认为,C3S体系早期水化速率降低是由于颗粒表面快速生成薄且连续的介稳水化硅酸钙层而引起的,这层水化产物被称作介稳态C-S-H (C-S-H(m))[15]。
C-S-H(m)通过阻碍未水化的C3S颗粒接触水,或通过阻碍离子由C3S颗粒表面扩散,从而抑制C3S体系的早期水化。在诱导期结束时,C-S-H(m)与溶液维持溶解平衡。
介稳层假说表明C-S-H(m)将C3S颗粒与溶液完全隔离,而且与溶液达到溶解平衡。但是,关于诱导期如何结束的机理则并不明晰。诱导期结束的准确时刻具有可重复性表明此时一定存在某种关键作用。换句话说,在诱导期结束时一定存在某种反应,使得C-S-H(m)最终变得不再稳定[15-16]。事实上,由图2可知,尽管诱导期内C3S水化放热速率大幅降低,但并不为零。核反应分析(nuclear resonance reaction analysis, NRRA)结果可知[17],随时间的推进氢元素逐渐侵入固相内部。氢元素侵入深度的增加表明,即使处于诱导期内,C3S的水化仍在进行。而且随着时间的推进,固相表面C-S-H的组成是变化的。

2.2 缓慢溶解假说(slow dissolution step hypothesis)
另一些学者认为,虽然介稳层假说可以解释部分实验结果,但仍存在一些重要的问题未解决。一方面,现今的技术并不能直接观察到C-S-H(m),如文献[22]所述,水泥颗粒表面存在蚀坑,其锋利的边缘使C-S-H(m)不太可能存在。另一方面,目前普遍认为最早生成的水化产物是C-S-H,但C-S-H的晶体结构与C3S的晶体结构完全不同,因此C-S-H在C3S表面很难致密附着,在颗粒表面多为孤立的团块存在[6],形貌如图3所示。

图3 水泥水化360 min后Alite颗粒表面上C-S-H的 SEM照片[6]Fig.3 SEM image of C-S-H on the surface of Alite grains after cement hydration for 360 min[6]
C3S水化进程中所形成的初始水化产物并不足以抑制C3S水化的进行。因此,Barret等[23-24]最早提出C3S表面与水接触生成表面羟基化层(superficially hydroxylated layer),表面羟基化的C3S其表观溶解度远小于C3S的理论溶解度,并且当溶液中CH浓度增加时,C3S的溶解进一步受到抑制。C3S溶出速率的结果表明[25],在没有C-S-H的形核和生长等因素的干扰下,溶液中钙元素和硅元素的浓度始终以3 ∶1的比例上升。因此,C3S体系水化诱导期的产生可以依据溶解-沉淀过程来解释,一些学者[7,26-29]在此基础上提出了缓慢溶解假说。
许多矿物相在水中的溶解速率与溶液的饱和状态间没有绝对的线性关系,换句话说,矿物相遇水后溶解速率下降是共价键矿物的常态[6,30-32]。新相的产生总是伴随着过冷或过饱和现象,需要一定的过冷度或过饱和度来提供最初形核的自由能,而在一定温度下自由能则是晶胚半径的函数[33]。反应相从光滑的颗粒表面溶解也同样需要额外的自由能来形成最初的溶蚀坑。图4为钠长石的溶解速率曲线,在稀溶液状态(即高度欠饱和状态)时,例如矿物相与液相接触的瞬间,液相中可溶解矿物相的浓度是极低的,此时矿物相的溶解速率很高,因为欠饱和溶液提供的自由能可以在矿物相表面形成最初的溶蚀坑[6-7,26-27,29]。而液相浓度高于某一阈值后,溶解速率急剧降低,因为液相缺少足够的自由能在矿物相表面继续溶蚀。而溶蚀晶体现有缺陷处所需的自由能低于溶蚀光滑的颗粒表面,因而溶解得以继续进行,但溶解速率下降。一般来说,形成溶蚀坑所需的自由能与伯氏矢量 (Burger’s vector) 有关。由于胶凝材料的晶体结构特点以及位错特性,晶体缺陷越多,诱导期越短[34-36]。Juilland等[22]使用SEM观察了不同条件下Alite颗粒的水化表面,发现在水中水化的Alite颗粒表面有明显的溶蚀坑,而在饱和CH溶液中水化的样品则保持了光滑的颗粒表面。该结果表明溶解假说可以为文献中的大量结果提供合理的解释,而且该过程也有试验结果的支持[37]。

图4 钠长石的溶解速率曲线[30]Fig.4 Dissolution rate curve of albite[30]
3 C3S体系的早期水化
3.1 C3S体系再次水化加速的原因
Skalny[15]列出了四种可能导致C3S体系再次水化加速的诱因,分别是C-S-H的形核和生长,稳定态C-S-H的生长,C-S-H(m)的破裂,以及CH的形核。
Bullard[38]使用模型模拟C3S体系水化进程中微观结构的发展以及溶液组成的变化,发现C-S-H的形核发生在C3S水化开始的前几分钟内。而且Bellmann等[18]的工作也说明C-S-H的生成先于C3S水化加速期的出现,因此,仅C-S-H的形核很难成为促进C3S体系再次水化加速的必要条件。
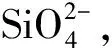
在C3S体系水化进程中,CH以及C-S-H晶体的沉淀和生长几乎同时进行。由于掺入CH可以延缓C3S体系的水化作用,因此CH的析出也曾被认为是C3S水化速率再次上升的原因之一。但是,在C3S水化样中掺入CH晶种并不会加速C3S体系的水化[39],甚至可能延缓加速期的出现[40]。同样,相比在水中进行C3S水化,CH溶液可以延缓C3S体系的水化进程[22]。因此,掺入少量的CH晶体并不能起到提供晶核的作用,因为CH晶体遇到水时会迅速溶解,溶液关于反应相的欠饱和度降低,从而降低C3S的溶解速率[22,41]。即使CH在溶液中快速饱和,由欠饱和度降低引起的反应相溶解速率下降将延缓C3S水化加速期的出现。随着液固比增加,CH的溶解度大于C-S-H,所以需要溶解更多C3S来析出CH。在此条件下,CH的沉淀是C3S加速溶解的结果,而不是导致其加速溶解的原因[42]。

3.2 C-S-H的生长与结构
目前,学者们确信C-S-H的生长与C3S体系的水化有关,而C-S-H的生长影响了所观察到的C-S-H结构。关于C-S-H结构的生长,存在以下两种理论,即纳米颗粒聚集理论 (aggregation of nanoparticles)[45-49],以及大而有缺陷的硅酸盐层理论 (large and defective sheets of silicate)[50]。
Gartner[50]提出了关于C-S-H的分支片状 (branching sheets) 生长机制,即硅氧四面体沿着二维方向形成聚集体,并且在这些层状硅酸盐结构中插入Ca2+以及—OH形成类似托贝莫来石 (tobermorite) 或六水硅钙石 (jennite) 的结构。C-S-H在形核后以该种方式生长,并且随着生长的进行,这些层状结构在纳米尺度上形成“短程有序”的原子排列。然而,随着层状结构的不断生长,由插层导致的点缺陷或线缺陷不断增多,这些生长缺陷引起晶体结构扭曲从而导致原子排列的“长程无序”,因此C-S-H在微观尺度上表现为晶体结构的无序化,也被称作C-S-H凝胶。
另一种C-S-H结构生长的理论是关于C-S-H纳米颗粒的聚集理论。该理论认为C-S-H固体颗粒仅能生长到几纳米便长时间保持稳定。然而,现有的C-S-H纳米颗粒可以促使新的颗粒在其表面形核,或者先前在溶液中形核的C-S-H颗粒在其表面聚集[44]。但是,新的形核过程要比在现有的C-S-H晶核表面继续生长所需的自由能更高 (即过饱和度更高),而且一些试验和模拟中,在高过饱和度状态下,只有水化最初的几分钟内可以出现C-S-H形核[21,26,41]。所以该理论可能存在一定的局限性。

图5 不同掺量下体系的 水化放热曲线[51]Fig.5 Heat flow curves of system hydration with different dosages of

5 结 语
猜你喜欢
上海金属(2022年4期)2022-08-03
中学生数理化·中考版(2022年12期)2022-02-16
云南化工(2021年9期)2021-12-21
铁道建筑技术(2019年6期)2019-11-29
钻井液与完井液(2019年4期)2019-10-10
精密成形工程(2018年6期)2018-11-23
材料工程(2017年7期)2017-07-25
中学化学(2016年10期)2017-01-07
分析科学学报(2016年2期)2016-10-15
中国学术期刊文摘(2016年8期)2016-02-13
