
新型吲哚类SIRT2 抑制剂的设计、合成及生物活性研究
2021-09-14 07:24王丽姣
西华大学学报(自然科学版) 2021年5期
王丽姣,宋 陈
(西华大学食品与生物工程学院,四川 成都 610039)
组蛋白修饰是蛋白翻译后修饰的重要修饰方式之一,包括乙酰化、甲基化、磷酸化修饰等。组蛋白乙酰化修饰受组蛋白乙酰化酶(HAT)和组蛋白去乙酰化酶(HDAC)共同调控,维持正常的生理功能。Surtuin(SIRT)蛋白,共包含7 种亚型,即SIRT1-SIRT7,是一种非典型去乙酰化酶,其催化底物去酰化机制与典型HDAC 不同,依赖NAD+的直接参与[1−3]。其中SIRT2 是一种存在于细胞质,但在有丝分裂期间可以穿梭于细胞核的重要亚型。有研究[4−6]表明,SIRT2 的异常与神经退行性疾病的发生、发展密切相关,因此,该靶点可作为相关疾病的潜在治疗靶点,SIRT2 抑制剂有望用于相应疾病的治疗。
1 已报道的SIRT2 抑制剂
许多科研小组报道了一系列的SIRT2 小分子抑制剂[7−8]。如图1 所示,根据其结构特征可将SIRT2 抑制剂分为多肽类底物类似物及非肽类小分子抑制剂两大类。多肽类抑制剂可能存在理化性质不好,易被代谢等成药性相对较差等缺点。非肽类小分子抑制剂又可分为以下9 类:烟酰胺类、羟基萘醛类、3-苯磺酰胺基苯基类、吲哚衍生物类、3,5-二取代-1,2,4-噁二唑、苯并吡喃酮、磺酰胺及磺酸类、氨基酸类、酰胺类等[9−10]。
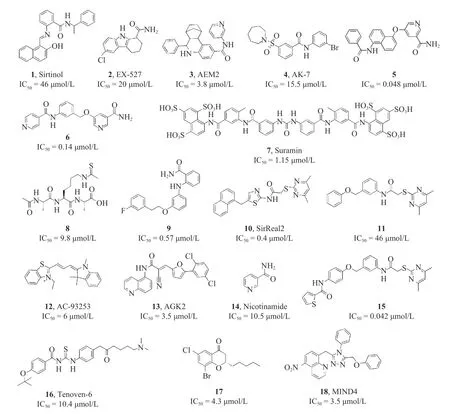
图1 已报道的部分SIRT2 抑制剂结构及IC50 值
目前,对SIRT2 小分子抑制剂的研究报道虽然较多,但大部分分子都面临着各种各样的问题,真正具有高活性、高选择性及理化性质好的SIRT2小分子抑制剂分子却较少。
本文前期研究获得了一个具有潜在的SIRT2抑活性的化合物N-(1-苄基-1H-吲唑-4-基)-2-(萘-1-基氧基)乙酰胺(14a),在100 和10 μmol·L−1浓度下,抑制率分别为70%和36%。本文从化合物14a 出发进行结构修饰,采用生物电子等排体原理,设计并合成了一系列新型吲哚类新骨架的目标化合物。
2 吲哚类目标化合物的设计
前期研究发现,当Linker 为2-氧基-乙酰胺,且芳香环为萘环时,化合物(14a)对SIRT2 的抑制活性有显著提高,在100 和10 μmol·L−1与阳性对照4 活性相当(73%,31%),抑制率分别为70%和36%。因此,本文在化合物14a 基础上进行改构,萘环和Linker2-氧基乙酰胺继续保留,根据生物电子等排体原理将吲唑环的2 位N 原子用C 原子代替而成吲哚环,然后再在吲哚环1-N 上引入疏水基团,从而设计合成了一系列新型吲哚类目标化合物(见图2)。

图2 目标化合物的设计
3 目标化合物的合成
吲哚类目标化合物的合成路线如图3 所示。由逆合成分析可知,合成目标化合物的其中一个原料为4-硝基吲哚,其与不同取代基的苄溴发生取代反应,与萘氧基乙酸缩合成酰胺,再将硝基还原成氨基得到不同取代胺的中间体。各种取代胺的中间体与另一个原料萘氧基乙酸缩合成不同的酰胺,最终得到不同的目标化合物。该设计路线不仅显著降低了实验成本,还提高了实验效率。因此,本文的合成路线即为先合成两个片段(即羧酸片段和氨基片段),再将其拼合成目标化合物。合成羧酸片段是用1-萘酚(19)和2-溴乙酸乙酯(20)在碳酸钾作敷酸剂,乙腈作溶剂下发生亲核反应得到化合物21,再将化合物21 在乙醇和水,碱性环境下水解生成中间体片段1-萘氧基乙酸(22)。4-硝基吲哚(23)与不同结构的疏水基团(苯环,带有取代基苯环,噻吩)24 同样在K2CO3作敷酸剂,N,N-二甲基甲酰胺(DMF)作溶剂下发生亲核反应得到化合物25,再经铁粉将硝基还原成氨基分别得到化合物26。

图3 吲哚类目标化合物27f-27o 的合成路线
羧酸化合物22 和游离的氨基化合物26 在2-(7-氮杂苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(HATU),N,N-二异丙基乙胺(DIEA),二氯甲烷(DCM)中缩合成最终目标化合物27f-27o。结构式如表1 所示。
最终合成了10 个目标化合物的结构如表1 所示,所有的化合物均通过了柱纯化或重结晶并用HPLC 鉴定了其纯度,纯度在95%以上,达到了测试生物活性的纯度要求,所有的目标化合物都进行1H-NMR、13C-NMR 和MS 的结构表征。

表1 化合物27f-27o 的结构
4 抑制活性测试与构效关系讨论
化合物27f-27o 的结构式如图4 所示。吲哚类化合物27f-27o 对SIRT2 的抑制活性数据分析见表2。

表2 吲哚类化合物27f-27o 对SIRT2 的抑制活性
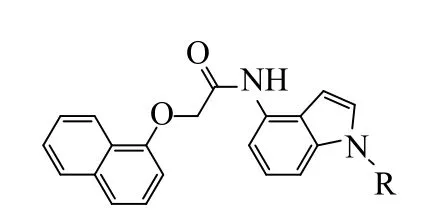
图4 化合物27f-27o
合成的10 个新分子对SIRT2 蛋白在高浓度下有一定的抑制效果,但总体的抑制率较低,均没有超过对照化合物14a。其中,所有化合物中抑制效果较好的是化合物27m 苯环间位溴双取代的结构,其在10 μmol·L−1下其抑制率只有42%,在100 μmol·L−1下为56%,可能是溴原子的电负性降低了苯环电子云密度,与SIRT2 的静电效应增强使得结合更牢固。其次是无取代的苯环的结构,其在10 μmol·L−1下其抑制率只有32%,在100 μmol·L−1下能达到55%,其余苯环上的单取代抑制率均在50%以下,噻吩取代的结构抑制率更低,可能仍然是共轭结构电子云效应以及取代基的诱导效应致使无法与SIRT2 蛋白结合或者结合不稳定。
5 合成实验操作及化合物表征数据
5.1 N-(1-(4-氟苄基)-1H-吲哚-4-基)-2-(萘-1-基氧基)乙酰胺(27f)的合成及表征
首先将1-萘酚19(10 g,69 mmol)用乙腈(30 mL 溶解在圆底烧瓶中,依次加入碳酸钾(28.6 g,207 mmol)、2-溴乙酸乙酯20(15 mL,138.7 mmol),加毕后升温至80 ℃下回流反应2 h 左右。经TLC检测反应完全后,过滤出不溶性杂质,浓缩滤液,经快速柱层析(PE)得白色雪花状固体化合物21。将中间体21(6.3 g,27.3 mmol)用乙醇(15 mL)和水(5 mL)溶解后,加入氢氧化钠(3.3 g,81.9 mmol),加完后继续室温下搅拌3 h 左右。经检测反应完全后,旋干乙醇溶剂,剩余水溶剂用稀盐酸调至弱酸性(PH=5 左右),此时析出固体,将其过滤,滤饼经真空干燥箱干燥后得到白色固体化合物22。两步收率40%。然后将4-硝基吲哚23(215 mg,1.32 mmol)、对氟溴苄24f(500 mg,2.65 mmol)、碳酸钾(546 mg,3.96 mmol)称入到反应瓶中,用N,N 二甲基甲酰胺DMF(10 mL)溶解后,升温至80 ℃下回流反应2 h 左右。经TLC 检测反应完全后加入水(100 mL),然后用乙酸乙酯萃取(50 mL×3),合并有机相,经无水硫酸钠干燥后,浓缩,经硅胶柱色谱分离(PE 与EA 体积比为50∶1),得黄色固体中间体25f。将中间体25f(445 mg,1.34 mmol)用乙醇(8 mL)和水(4 mL)溶解在反应瓶中,一次性加入还原铁 粉(376 mg,6.72 mmol)、氯 化 铵(36 mg,0.67 mmol),加毕后升温至80 ℃下回流1 h 左右。经TLC 检测反应完全后,趁热过滤出不溶性杂质,旋干滤液中的乙醇,加入少量水后用饱和碳酸氢钠溶液将PH 调至8,然后用乙酸乙酯萃取(30 mL×3),合并有机相,用无水硫酸钠干燥后,减压浓缩旋干后得红棕色固体化合物26f。将中间体22(151 mg,0.75 mmol)和中间体26f(150 mg,0.62 mmol)称入反应瓶中,用乙腈溶解后依次加入2-(7-氮杂苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(HATU)(283 mg,0.75 mmol),N,N-二异丙基乙胺(DIEA)(205 μL,1.24 mmol),加毕后继续常温下搅拌反应3 h 左右。经TLC 检测反应完全后,旋干溶剂,加水和乙酸乙酯萃取(30 mL×3),合并有机相并用无水硫酸钠干燥后,减压浓缩,经硅胶柱色谱分离(PE∶EA 体积比为2∶1),得目标化合物27f,红色固体,总收率为40%,纯度为98.1%。1H NMR(400 MHz,DMSO-d6) δ 9.93 (s,1H),8.41-8.39 (m,1H),7.96 —7.94 (m,1H),7.66 —7.56 (m,4H),7.53—7.46 (m,2H),7.32—7.27 (m,3H),7.20—7.10(m,3H),7.05 (d,J=7.6 Hz,1H),6.73 (d,J=2.8 Hz,1H),5.45 (s,2H),5.09 (s,2H) ppm.13C NMR (101 MHz,DMSO-d6) δ 166.86,163.11,160.69,153.96,136.99,134.87,134.61,130.53,129.52,128.69,127.93 1,27.04 1,26.52 1,25.87 1,25.41 1,22.31 1,21.96 1,21.72,121.07,115.88,115.66,112.00,107.31,105.99,99.48,67.99,48.95 ppm.HRMS:m/z calcd for C27H21FN2O2[M+H]+425.1660,found 425.1656。
5.2 N-(1-(4-氯苄基)-1H-吲哚-4-基)-2-(萘-1-基氧基)乙酰胺(27g)的合成及表征
将4-硝基吲哚23(197 mg,1.20 mmol)、对氯溴苄24g(500 mg,2.40 mmol)、碳酸钾(497 mg,3.60 mmol)称入到反应瓶中,用N,N 二甲基甲酰胺DMF(10 mL)溶解后,升温至80 ℃下回流反应2 h 左右。经TLC 检测反应完全后加入水(100 mL),经乙酸乙酯萃取(50 mL×3),有机层经无水硫酸钠干燥后,浓缩,经硅胶柱色谱分离(PE 与EA 体积比为5∶1)得黄色固体中间体25g。将中间体25g(400 mg,1.40 mmol)用乙醇(8 mL)和水(4 mL)溶解在反应瓶中,一次性加入还原铁粉(391 mg,6.98 mmol)、氯化铵(37 mg,0.70 mmol),加毕后升温至80 ℃下回流1 h 左右。经TLC 检测反应完全后,趁热过滤出不溶性杂质,旋干滤液中的乙醇,加入少量水后用饱和碳酸氢钠溶液将PH 调至8,然后用乙酸乙酯萃取(30 mL×3),合并有机相并用无水硫酸钠干燥后,减压浓缩旋干后得红棕色固体化合物26g。将中间体22(142 mg,0.70 mmol)和中间体26g(150 mg,0.58 mmol)称入反应瓶中,用乙腈溶解后依次加入2-(7-氮杂苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(HATU)(266 mg,0.70 mmol),N,N-二异丙基乙胺(DIEA)(192 μL,1.16 mmol),加毕后继续常温下搅拌反应3 h 左右。经TLC 检测反应完全后,类似合成27f 后处理及硅胶柱色谱分离后得目标化合物27g,红色固体,总收率为45%,纯度为98.2%。1H NMR (400 MHz,DMSO-d6) δ 9.91 (s,1H),8.38—8.36 (m,1H),7.92—7.89 (m,1H),7.62 (d,J=8.0 Hz,1H),7.58—7.52 (m,3H),7.48 (d,J=3.2 Hz,1H),7.45 (t,J=8.0 Hz,1H),7.39—7.35 (m,2H),7.25—7.18 (m,3H),7.08 (d,J=8.0 Hz,1H),7.01 (d,J=7.6 Hz,1H),6.71(d,J=2.8 Hz,1H),5.43 (s,2H),5.06 (s,2H) ppm.13C NMR (101 MHz,DMSO-d6) δ 166.87,153.97,137.71,137.01,134.61,132.43,130.56,129.27,128.98,128.76,127.93,127.04,126.52,125.87,125.42,122.32,122.01,121.72,121.07,112.04,107.28,105.99,99.57,68.00,48.98 ppm.HRMS:m/z calcd for C27H21ClN2O2[M+H]+441.1364,found 441.1363。
5.3 N-(1-(4-溴苄基)-1H-吲哚-4-基)-2-(萘-1-基氧基)乙酰胺(27h)的合成及表征
将4-硝基吲哚23(162 mg,1.00 mmol)、对溴溴苄24h(500 mg,2.00 mmol)、碳酸钾(414 mg,3.00 mmol)称入到反应瓶中,用N,N 二甲基甲酰胺DMF(10 mL)溶解后,升温至80 ℃下回流反应2 h 左右。经TLC 检测反应完全后经类似后处理及硅胶柱色谱分离得黄色固体中间体25h。后续步骤同合成27f 类似,得目标化合物27h,黄色固体,总收率为40%,纯度为98.1%。1H NMR (400 MHz,DMSO-d6) δ 9.91 (s,1H),8.38—8.36 (m,1H),7.92—7.89 (m,1H),7.62 (d,J=7.2 Hz,1H),7.58—7.42 (m,7H),7.23 (d,J=8.0 Hz,1H),7.13 (d,J=8.4 Hz,2H),7.08 (t,J=8.0 Hz,1H),7.01 (t,J=8.0 Hz,1H),6.71 (d,J=2.8 Hz,1H),5.41 (s,2H),5.06(s,2H) ppm.13C NMR (101 MHz,DMSO-d6) δ 166.87,153.97,138.13,137.01,134.61,131.90,130.57,129.61,128.76,127.93,127.04,126.52,125.87,125.42,122.32,122.02,121.72,121.07,120.92,112.04,107.27,105.99,99.58,68.00,49.04ppm.HRMS:m/z calcd for C27H21BrN2O2[M+H]+485.0859,found 485.085 2。
5.4 N-(1-(4-乙酰基苄基)-1H-吲哚-4-基)-2-(萘-1-基氧基)乙酰胺(27i)的合成及表征
将4-硝基吲哚23(190 mg,1.17 mmol)、1-(4-(溴甲基)苯基)乙-1-酮24i(500 mg,2.35 mmol)、碳酸钾(484 mg,3.51 mmol)称入到反应瓶中,用N,N 二甲基甲酰胺DMF(10 mL)溶解后,升温至80℃下回流反应2 h 左右。经TLC 检测反应完全后经类似萃取及硅胶柱色谱分离得黄色固体中间体25i。后续步骤同合成27f 类似,得目标化合物27i,总收率为42%,纯度为98.2%。1H NMR (400 MHz,DMSO-d6) δ 9.93 (s,1H),8.37—8.36 (m,1H),7.89(d,J=8.0 Hz,3H),7.63—7.43 (m,6H),7.28 (d,J=8.0 Hz,2H),7.22 (d,J=8.0 Hz,1H),7.07 (t,J=8.0 Hz,1H),7.02 (d,J=7.2 Hz,1H),6.73 (d,J=2.4 Hz,1H),5.53 (s,2H),5.06 (s,2H),2.53 (s,3H) ppm.13C NMR (101 MHz,DMSO-d6) δ 197.89,166.88,153.98,143.97,137.09,136.45,134.61,130.59,129.00,128.90,127.93,127.48,127.04,126.53,125.87,125.42,122.32,122.04,121.74,121.07,112.08,107.27,105.99,99.65,68.00,49.43,27.13 ppm.HRMS:m/z calcd for C29H24N2O3[M+H]+449.1860,found 449.1862。
5.5 N-(1-(4-甲氧基苄基)-1H-吲哚-4-基)-2-(萘-1-基氧基)乙酰胺(27j)的合成及表征
将4-硝基吲哚23(202 mg,1.24 mmol)、1-(溴甲基)-4-甲氧基苯24j(500 mg,2.49 mmol)、碳酸钾(513 mg,3.72 mmol)称入到反应瓶中,用N,N 二甲基甲酰胺DMF(10 mL)溶解后,升温至80 ℃下回流反应2 h 左右。经TLC 检测反应完全后经类似萃取和硅胶柱色谱分离得黄色固体中间体25j。后续步骤同合成27f 类似,得目标化合物27j,总收率为34%,纯度为98.1%。1H NMR (400 MHz,DMSO-d6) δ 9.88 (s,1H),8.37 —8.35 (m,1H),7.92—7.89 (m,1H),7.61—7.52 (m,4H),7.46—7.42(m,2H),7.27 (d,J=8.0 Hz,1H),7.17 (d,J=7.2 Hz,2H),7.07 (t,J=8.0 Hz,1H),7.01 (d,J=7.6 Hz,1H),6.87 (d,J=8.8 Hz,2H),6.66 (d,J=2.8 Hz,1H),5.33 (s,2H),5.05 (s,2H),3.70 (s,3H) ppm.13C NMR(101 MHz,DMSO-d6) δ166.83,159.05,153.97,136.99,134.61,130.52,130.48,128.95,128.63,127.93,127.04,126.53,125.87,125.41,122.31,121.80,121.06,114.39,111.85,107.39,105.99,99.20,68.00,55.53,49.24ppm.HRMS:m/z calcd for C28H24N2O3[M+H]+437.1860,found 437.1868。
5.6 N-(1-(4-氰基苄基)-1H-吲哚-4-基)-2-(萘-1-基氧基)乙酰胺(27k)的合成及表征
将4-硝基吲哚23(207 mg,1.28 mmol)、4-(溴甲基)苄腈24k(500 mg,2.55 mmol)、碳酸钾(528 mg,3.83 mmol)称入到反应瓶中,用N,N 二甲基甲酰胺DMF(10 mL)溶解后,升温至80 ℃下回流反应2 h 左右。经TLC 检测反应完全后经类似萃取,硅胶柱色谱分离得黄色固体中间体25k。后续步骤同合成27f 类似,得目标化合物27k,总收率为43%,纯度为98.4%。1H NMR (400 MHz,DMSOd6) δ 9.94 (s,1H),8.37—8.35 (m,1H),7.91—7.89(m,1H),7.79—7.76 (m,2H),7.62 (d,J=7.6 Hz,1H),7.58—7.51 (m,4H),7.44 (t,J=8.0 Hz,1H),7.29 (d,J=8.4 Hz,2H),7.21 (d,J=8.0 Hz,1H),7.07 (t,J=8.0 Hz,1H),6.99 (d,J=8.0 Hz,1H),6.73 (d,J=2.8 Hz,1H),5.55 (s,2H),5.06 (s,2H) ppm.HRMS:m/z calcd for C28H21N3O2[M+H]+432.1707,found 432.1703。
5.7 N-(1-(4-甲基苄基)-1H-吲哚-4-基)-2-(萘-1-基氧基)乙酰胺(27l)的合成及表征
将4-硝基吲哚23(102 mg,1.24 mmol)、1-(溴甲基)-4-甲基苯24l(500 mg,2.49 mmol)、碳酸钾(513 mg,3.72 mmol)称入到反应瓶中,用N,N 二甲基甲酰胺DMF(10 mL)溶解后,升温至80 ℃下回流反应2 h 左右。经TLC 检测反应完全后经类似萃取,硅胶柱色谱分离得黄色固体中间体25l。后续步骤同合成27f 类似,得目标化合物27l,总收率为41%,纯度为98.3%。1H NMR (400 MHz,DMSOd6) δ 9.90 (s,1H),8.38—8.36 (m,1H),7.93—7.90(m,1H),7.62 (d,J=8.0 Hz,1H),7.59—7.52 (m,3H),7.46—7.43 (m,2H),7.24 (d,J=8.0 Hz,1H),7.14—7.05 (m,5H),7.01 (d,J=8.0 Hz,1H),6.69 (d,J=2.4 Hz,1H),5.37 (s,2H),5.06 (s,2H),2.25 (s,3H)ppm.13C NMR (101 MHz,DMSO-d6) δ 166.84,153.98,137.06,136.99,135.59,134.61,130.49,129.52,128.75,127.93,127.48,127.04,126.53,125.87,125.42,122.32,121.83,121.07,111.87,107.36,106.00,99.25,68.01,49.54,21.10 ppm.HRMS:m/z calcd for C28H24N2O2[M+H]+421.1911,found 421.1905。
5.8 N-(1-(3,5-二溴苄基)-1H-吲哚-4-基)-2-(萘-1-基氧基)乙酰胺(27m)的合成及表征
将4-硝基吲哚23(74 mg,0.46 mmol)、1,3-二溴-5-(溴甲基)苯24m(300 mg,0.91 mmol)、碳酸钾(190 mg,1.38 mmol)称入到反应瓶中,用N,N 二甲基甲酰胺DMF(10 mL)溶解后,升温至80 ℃下回流反应2 h 左右。经TLC 检测反应完全后加入水(100 mL),然后用乙酸乙酯萃取(50 mL×3),合并有机相,经无水硫酸钠干燥后,浓缩,经硅胶柱色谱分离(PE 与EA 体积比为5:1)得黄色固体中间体25m。后续步骤同合成27f 类似,得目标化合物27m,总收率为41%,纯度为98.0%。1H NMR (400 MHz,DMSO-d6) δ 9.93 (s,1H),8.43—8.31 (m,1H),7.90 (d,J=9.4 Hz,1H),7.73 (t,J=1.6 Hz,1H),7.63 (d,J=7.6 Hz,1H),7.58—7.51 (m,4H),7.45 (t,J=7.9 Hz,1H),7.39 (d,J=1.6 Hz,2H),7.29 (d,J=8.2 Hz,1H),7.12 (d,J=7.9 Hz,1H),7.01 (d,J=7.6 Hz,1H),6.72 (d,J=3.0 Hz,1H),5.45 (s,2H),5.05 (s,2H).13C NMR (101 MHz,DMSO-d6)) δ 166.89,153.97,143.51,136.95,134.60,132.83,130.63,129.40,128.76,127.92,127.03,126.53,125.86,125.41,123.07,122.33,122.26,121.06,112.26,107.16,106.00,99.98,67.98,48.35 ppm.HRMS:m/z calcd for C27H20BrN2O2[M+H]+562.9964,found 562.9975。
5.9 N-(1-苄基-1H-吲哚-4-基)-2-(萘-1-基氧基)乙酰胺(27n)的合成及表征
将4-硝基吲哚23(237 mg,1.46 mmol)、卞溴24n(500 mg,2.92 mmol)、碳酸钾(604 mg,4.38 mmol)称入到反应瓶中,用N,N 二甲基甲酰胺DMF(10 mL)溶解后,升温至80 ℃下回流反应2 h 左右。经TLC 检测反应完全后加入水(100 mL),然后用乙酸乙酯萃取(50 mL×3),合并有机相,经无水硫酸钠干燥后,浓缩,经硅胶柱色谱分离(PE 与EA 体积比为5∶1)得黄色固体中间体25n。后续步骤同合成27f 类似,得目标化合物27n,总收率为45%,纯度为98.3%。1H NMR (400 MHz,DMSOd6) δ 9.24 (s,1H),8.37—8.35 (m,1H),7.91—7.89(m,1H),7.62 (d,J=7.6 Hz,1H),7.56—7.53 (m,3H),7.48 (d,J=3.2 Hz,1H),7.43 (t,J=8.0 Hz,2H),7.31—7.17 (m,6H),7.06 (t,J=8.0 Hz,1H),6.99 (d,J=8.0 Hz,1H),6.70 (d,J=2.8 Hz,1H),5.42 (s,2H),5.05 (s,2H) ppm.HRMS:m/z calcd for C27H22N2O2[M+H]+407.1754,found 407.1749。
5.10 2-(萘-1-基氧基)-N-(1-(噻吩-2-基甲基)-1H-吲哚-4-基)乙酰胺(27o)的合成及表征
将2-噻吩甲醇(500 mg,4.38 mmol)用二氯甲烷(10 mL)溶解在反应瓶中,缓慢加入二氯亚砜(1.5 mL,21.9 mmol)继续常温下搅拌1 h 左右。经TLC 检测反应完全后,旋干溶剂,得中间体粗产品2-氯噻吩。将4-硝基吲哚23(309 mg,1.90 mmol)、2-氯噻吩(500 mg,3.80 mmol)、碳酸钾(787 mg,5.70 mmol)称入到反应瓶中,用N,N 二甲基甲酰胺DMF(10 mL)溶解后,升温至80 ℃下回流反应2 h 左右。经TLC 检测反应完全后经相似后处理得黄色固体中间体25o。后续步骤同合成27f 类似,得目标化合物27o,红色固体,总收率34%,纯度为98.2%。1H NMR (400 MHz,DMSO-d6) δ 9.90(s,1H),8.37—8.35 (m,1H),7.91—7.89 (m,1H),7.72 (t,J=2.4 Hz,1H),7.62 (d,J=7.6 Hz,1H),7.58—7.52 (m,4H),7.44 (d,J=8.0 Hz,1H),7.38(d,J=1.6 Hz,2H),7.28 (d,J=8.0 Hz,1H),7.10 (t,J=8.0 Hz,2H),7.00 (d,J=7.6 Hz,1H),6.72 (d,J=3.2 Hz,1H),5.61 (s,2H),5.05 (s,2H) ppm.13C NMR(101 MHz,DMSO-d6) δ 166.85,153.98,141.13,136.84,134.61,130.54,128.22,127.93,127.33,127.04,126.80,126.53,126.21,125.87,125.42,122.32,121.95,121.70,121.07,112.06,107.27,106.00,99.69,68.00,44.61 ppm.HRMS:m/z calcd for C25H20N2O2S [M+H]+413.1318,found 413.1305。
6 总结与展望
本文以化合物14a 为先导化合物,根据生物电子等排体原理将吲唑环的2-位N 原子用C 原子代替而成吲哚环,然后再在吲哚环1-N 上引入疏水基团,从而合成了10 个新型吲哚类目标化合物。其中,抑制效果较好的是化合物27m,其在10 μmol·L−1下其抑制率为42%,在100 μmol·L−1下为56%,这可能是溴原子的电负性降低了苯环电子云密度,与SIRT2 的静电效应增强使得结合更牢固。本文通过合理药物设计并合成了一系列全新SIRT2 抑制剂,通过酶水平活性评价获得了较好抑制活性的化合物27m,为靶向SIRT2 的抗肿瘤药物研究提供了更多的SIRT2 新骨架选择,并为课题后期进一步的结构修饰奠定了基础。
猜你喜欢
中国药理学与毒理学杂志(2022年7期)2022-10-17
中国药学药品知识仓库(2022年10期)2022-05-29
中国典型病例大全(2022年10期)2022-05-10
动物营养学报(2022年1期)2022-02-20
中国应急管理科学(2021年9期)2021-03-16
人物画报(2020年29期)2020-03-14
人物画报(2020年36期)2020-03-13
山东工业技术(2018年9期)2018-05-26
股市动态分析(2015年12期)2015-09-10
中小企业管理与科技·中旬刊(2014年10期)2015-02-03
