
重复交叉设计生物等效性研究样本量计算的确切法与随机模拟法
2022-01-19 08:40王学文袁加盟王洪源
中国卫生统计 2021年6期
单 雪 王 登 王学文 袁加盟 王洪源
【提 要】 目的 研究重复交叉设计临床试验平均生物等效性评价样本量计算的确切法与随机模拟法的统计学特性,推荐选择较优的分析方法。方法 系统的介绍确切法与采用参比制剂校正随机模拟样本量计算原理及步骤,采用计算机模拟技术,比较两种方法在不同情境下的表现。结果 在高变异药物常见应用情境下,不同参数设定下通过确切法进行样本量计算的结果小于采用随机模拟估计的样本量,且随着CVWR%增大,或几何均值比值偏离1,两者的样本量差距增大。结论 确切法与随机模拟法样本量计算过程均根据参比制剂个体内变异(CVWR%)对置信区间进行放宽,但由于确切法采用的是CVWR%预计值而不是实际取值,且未考虑对点估计值的等效要求等缺点,建议优先选择随机模拟方法估计高变异药物等效评价样本量。
生物等效性(bioequivalence,BE)研究是比较受试制剂(T)与参比制剂(R)的药代动力学(PK)参数,即评价药物吸收速度和吸收程度差异是否在可接受范围内的研究,一般建议采用2制剂2周期交叉设计的方法。根据受试/参比制剂主要PK参数几何均值比值(geometric mean ratio,GMR)的90%置信区间评价,等效标准为(80.00%,125.00%)[1]。
某些药物由于吸收、代谢、稳定性等原因,导致一个或多个PK参数的个体内变异系数(within-subject coefficient of variation,CVW%)大于或等于30%,称为高变异药物(highly variable drug,HVD)[2]。对于安全性较好的高变异药物,PK参数等效评价采用参比制剂校正的生物等效性(reference-scaled average bioequivalence,RSABE)方法[3]。与2×2设计非校正等效评价的区别是RSABE等效标准随CVWR%实际取值变化,需要采用高阶的交叉设计,即重复交叉设计,允许在80.00%~125.00%基础上放宽等效界值,同时要求GMR在80.00%~125.00%内[2]。
目前为止,已有学者发表了高阶重复交叉设计样本量估算方式,Phani等基于2×2交叉设计的近似样本量公式[4]、刘甜甜等的确切法[5]、随机模拟估计方式[6]。本研究将以高变异药物生物等效性评价试验为例,详细介绍重复交叉设计样本量计算的随机模拟技术,并与确切法进行统计特性比较。
样本量估计方法
1.高变异药物生物等效性评价样本量计算的确切法
在试验设计阶段,通过既往资料估计参比制剂的个体内标准差SWR,根据以下公式求得经调整的等效性界值,通过迭代计算相应的2制剂2序列2周期试验设计的样本量,再乘以固定系数,得到2制剂3序列3周期部分重复设计或2制剂2序列4周期完全重复设计的样本量[4-5]。等效界值调整公式如下:
其中,SW0=0.25,为法规规定的常数值。
得到2×2设计下的样本量后,2×3×3(部分重复设计)样本量为上述样本量乘以系数3/4,2×2×4(完全重复设计)样本量为上述样本量乘以系数1/2。
2.高变异药物生物等效性评价样本量计算的随机模拟方法
决定高变异药物等效评价样本量的参数,包括参比制剂的个体内标准差SWR,受试制剂的个体内标准差SWT,受试/参比几何均值比值GMR,以及1类错误率α及2类错误率β。采用直接模拟样本参数形式产生随机样本[7-10],判断是否成功(即等效),相同预计参数值的100000个随机模拟样本的成功率为统计效能,通过迭代得到预设统计效能的最小样本量。
具体模拟步骤如下:
(1)设定迭代起始样本量,按照如下方式生成nsims=100000个随机模拟样本,每个随机模拟样本主要由以下参数定义:


表1 与试验设计相关的E(mse)计算公式


④产生nsims个符合卡方分布的SWR2的随机数据:参比制剂个体内均方差SWR2=σWR2×rchi(nisms,dfRR)/dfRR,服从自由度为dfRR的卡方分布,SWR为样本取值,即每一个随机模拟情境下或单个样本的实际取值,σWR为估计的SWR真实值。

(3)符合相应标准则将结论计为“成功”,否则,计为“失败”,并计算成功的百分比,即为在相应样本量和参数设置情况下的统计效能。
(4)若该检验效能大于等于设定的目标统计效能,则计算过程结束。否则样本量加一个最小单位(通常为给药序列数的整数倍,如3序列半重复设计为3,2序列完全重复设计为2),继续重复步骤2.1 至2.4直至达到停止标准。
(5)模拟结束,输出达到目标统计效能的最小样本量。
模拟评价及样本量计算结果比较
采用高变异药物生物等效性评价在实际应用中的常见情境及参数设置进行随机模拟实验。试验设计采用2制剂3序列3周期,设定检验水平为单侧0.05,统计效能为80%,不同GMR和CVW估计值对应的随机模拟估计样本量和采用确切方法估计样本量比较趋势如图1所示。表2提供了部分具体值,并列出了两种方法计算的样本量的差值,同时提供了(0.8,1.25)根据SWR调整后的等效性界值。可以看到,在高变异(CVW≥0.3)范围内随机模拟估计的样本量均不同程度的大于确切法估计的样本量,且随着GMR偏离1,或CVW增大,样本量差异呈扩大的趋势。

表2 2制剂3序列3周期部分重复交叉设计随机模拟样本量估算与确切方法比较

图1 在不同GMR的情况下确切法和随机模拟方法所需样本量随CV变化的趋势图,CVW=0.3为高变异(CVW≥0.3)标准分隔线
当等效评价标准固定时,即非高变异药物,达到一定统计效能,所需要的样本量,与GMR和个体内变异的关系呈固定规律。而采用参比制剂校正方法时,由于评价方法的复杂和等效界值的不确定,在不同GMR时,样本量并不随CV单调变化。
法规要求高变异药物受试制剂与参比制剂PK参数几何均值比值点估计值在80~125内。图2给出了不同GMR预计值及CV预计值的情况,随机模拟实际数据GMR落在限定范围外的概率。可以看到,同一GMR预计值的情况下,随着CV增大,GMR随机模拟实际值不符合等效要求的概率逐渐增大。同一CV预计值的情况下,GMR预计值越偏离1,GMR随机模拟实际值不符合等效要求的概率显著增大。GMR预计值及CV预计值对实际GMR落在限定范围外的影响具有叠加效应。
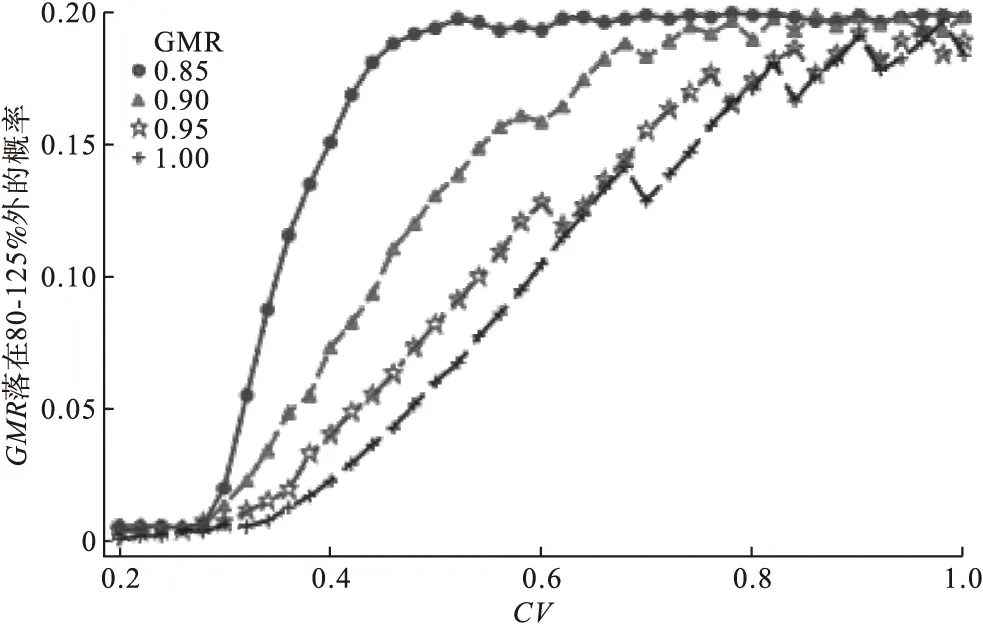
图2 在不同GMR的情况下随CV增大GMR落在80~125%外的概率变化趋势
讨 论
高阶重复交叉设计由于采用自身对照,可以估计特定制剂的个体内变异,在高变异药物及窄治疗指数药物等效评价中得到广泛应用[11-13]。Phani等[4]提出一种进行间接估计的方法,首先采用SWR计算校正后等效界值,带入2×2交叉设计的近似公式计算样本量,然后,2×3×3(部分重复设计)为上述样本量乘以系数3/4,2×2×4(完全重复设计)为上述样本量乘以系数1/2。2×2交叉设计的样本量计算公式本身是一种基于较大样本假设的近似。刘甜甜等[5]采用Meiyu Shen等[14]提出的迭代方式计算确切样本量的过程替代了公式近似估计2×2样本量计算过程,然后同样乘以固定系数估计重复交叉设计样本量,对Phani方法进行了一定改进。此外,最近还有学者提出调整参数后利用PASS软件的2×2样本量计算过程进行计算的方式[15],估计结果与刘甜甜提出的方法相似。Laszlo等[6]采用模拟受试者制剂水平数据的随机模拟方式计算样本量。本文详细介绍了模拟样本水平参数的方式进行样本量估计的方法,并提供了高变异药物生物等效性评价实际应用场景下,对确切法及随机模拟方法的比较。
高变异药物PK参数的变异较大,同时生物等效试验有小样本的特点,这使得GMR和CVW的不确定性增大,对是否等效起决定作用[16]。确切方法对于生物等效判断标准进行了过度的简化。首先,随着GMR预计值偏离1,CV预计值增大,GMR实际值落在80%~125%以外的概率不能忽视。其次,试验设计阶段预估的SWR往往来自历史数据,试验结束后计算的SWR实际取值存在不确定性,尤其是计划阶段SWR预估值在0.3附近时,有相当的概率小于0.294而导致选择ABE,使得统计效能显著低于预期,即使选择RSABE方法,具体等效界值也是不确定的。采用随机模拟方式计算样本量,由于随机模拟SWR的分布,而不是采用固定值,在很大程度上降低了SWR取值不确定的风险。本研究样本量计算及随机模拟均采用SAS软件完成,对不同试验设计下生物等效评价样本量计算(FDA及EMA标准略有不同,CDE评价标准同FDA)均已形成网络操作界面工具。
猜你喜欢
内蒙古统计(2021年4期)2021-12-06
今日农业(2020年18期)2020-12-14
初中生世界·八年级(2019年6期)2019-08-13
测控技术(2018年4期)2018-11-25
上海精神医学(2017年5期)2017-11-29
中成药(2017年4期)2017-05-17
小学生导刊(低年级)(2016年9期)2016-10-13
小学生导刊(低年级)(2016年6期)2016-07-02
中国中医药现代远程教育(2014年11期)2014-08-08
振动、测试与诊断(2014年6期)2014-03-01
