
聚甘露糖醛酸胶束的制备及其载药性能
2022-01-20 10:59孙家曦王腾斌陈祥艳张永勤
青岛科技大学学报(自然科学版) 2022年1期
孙家曦,王腾斌,陈祥艳,张永勤*
(1.中国海洋大学 海洋药物教育部重点实验室;山东省糖科学与糖工程重点实验室,山东 青岛 266003;2.青岛科技大学 化工学院,山东 青岛 266042)
褐藻胶来源于海洋资源极为丰富的褐藻,其钠 盐被称为海藻酸钠,具有廉价、易成凝胶、生物相容性、无毒性、可生物降解等优点[1],被广泛应用于医药行业中,如伤口敷料[2]、水凝胶[3]、水凝胶微珠[4]、止血剂[5]等。它是由α-L-古罗糖醛酸(G)及β-D-甘露糖醛酸(M)通过1-4糖苷键连接而成[6]。在褐藻胶的糖链上,G和M无序排列,从而构成聚甘露糖醛酸连续片段(PM)、聚古罗糖醛酸连续片段(PG)和甘露糖醛酸与古罗糖醛酸的交替连接片段[7]。甘露糖醛酸和古罗糖醛酸两种糖醛酸的结构非常相似,其差别仅在于C5上的羟基位置不同,但是,它们聚合成链后的空间构象差别却非常大,PG具有结构刚性,易与二价阳离子形成蛋壳结构,并以此影响胶束的形成以及降低所包载药物的释放速率,而PM具有结构柔性,更易在溶液中自组装形成胶束结构,也因此决定了其理化性质的巨大差异[8-11]。聚甘露糖醛酸是由褐藻胶经部分酸解后,利用PM和PG在酸性环境中溶解度不同进行分级沉淀获得[12],具有抗氧化[13]、免疫调节[14]、抗阿尔兹海默[15]等多种生物活性。PM结构中无古罗糖醛酸,因而其性降低,水溶性增大,结构柔性增强,有望成为胶束结构中理想的亲水嵌段,为PM在药剂辅料领域开辟新的应用方向。
PM结构中所含有的羧酸基团具有较强的反应活性,可利用该基团引入其它疏水基团,制成两亲性胶束材料,用于包载疏水性药物,以提高该类药物的靶向性并降低其副作用。迄今为止,PM在递药系统中的应用较少,丛萌逸等[16]将油胺直接接枝于PM制成胶束,用于治疗过敏性结膜炎,然而,PM在抗肿瘤药物中的应用尚未见报道。脱氧胆酸(DA)作为一种疏水性分子,常用于抗肿瘤两亲性胶束材料的制备[17-20],其结构中具有较强活性的羧酸基团可通过双功能基团乙二胺接枝于多糖分子。然而,由于PM结构中的每个糖单元均含有羧酸基团,按照文献普遍采用的方法制备[21-22],即,以PM为合成起始原料,得到的聚甘露糖醛酸-乙二胺-脱氧胆酸(PM-cc-DA)胶束材料的难度较大。究其原因,乙二胺极易使PM发生分子间和分子内交联,即便未发生交联,在后续接枝脱氧胆酸反应中,也很难保证乙二胺另一端的裸露氨基均与脱氧胆酸反应,这将对后续的胶束制备造成不利影响。为解决上述问题,本研究拟以脱氧胆酸为起始合成原料,首先制成脱氧胆酸-乙二胺(DA-EDA),然后再将其接枝于PM。为保证PM-cc-DA的质量,对DA-EDA的纯度要求至关重要,目前尚未见此方面的报道。因此,本研究拟首先研究DA-EDA的分离纯化方法,并合成PM-cc-DA,再以抗肿瘤药物阿霉素(DOX)作为模型药物,考察其载药胶束的载药性能。
1 实验部分
1.1 试剂与仪器
聚甘露糖醛酸,中国海洋大学医药学院糖工程与制剂实验室;脱氧胆酸,美国sigma公司;N,N-二异丙基碳二亚胺、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、N-羟基丁二酰亚胺、盐酸阿霉素,上海阿拉丁生化科技股份有限公司;乙二胺、N,N-二甲基甲酰胺、甲酰胺,国药试剂有限公司;透析袋(MWCO 2000)。
核磁共振波谱仪,Pro pulse 500MHz型,安捷伦科技(美国)有限公司;紫外可见分光光度计,UV-6000PC型,上海元析仪器有限公司;Molecular Devices EMax Plus®酶标仪,美谷分子仪器(上海)有限公司;冷冻干燥机,FD-1-50型,北京博医康实验仪器有限公司;真空干燥箱,PCD-C3000 Series型,合肥达斯卡特生物科技有限公司;便携式电导率仪,DDB-303 A型,上海仪电科学仪器股份有限公司。
1.2 氨基化脱氧胆酸(DA-EDA)的制备与表征
将乙二胺(EDA)4.5 g溶于15 m L甲酰胺溶液中,配成EDA溶液。在冰浴条件下,将脱氧胆酸(DA)3.92 g溶于10 m L无水N,N-二甲基甲酰胺(DMF)中,加入1.27 gN-羟基丁二酰亚胺(NHS),密封抽真空后充入氮气,加入1.7 m LN,N-二异丙基碳二亚胺(DIC),活化4 h。将该混合液于-20℃冰箱静置以促进晶体析出,过膜,取滤液,逐滴加入到EDA溶液中,反应6 h。将反应液滴入水溶液中进行沉淀,离心后加入酸水溶解后采用乙酸乙酯进行萃取以除去杂质,获得产物水溶液,冷冻干燥后获得DA-EDA固体。
1.3 PM-cc-DA的制备与表征
将0.4 g PM溶于300μL水中,加入25 m L甲酰胺、0.24 g NHS及0.44 g 1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC),充分混匀使其溶解,密封并抽真空再充氮,活化4 h。将DA-EDA的DMF溶液(制备高中低三种不同取代度PM-cc-DA所用DA-EDA分别为0.434、0.217以及0.108 g),逐滴加入到上述活化的PM多糖溶液中,反应12 h。将该反应液滴加至95%乙醇中醇沉,于4℃冰箱静置,在3 000 r·min-1下离心,得沉淀。将沉淀用95%乙醇洗涤后溶解于水及甲酰胺混合液(1∶1)中,用去离子水透析24 h,冷冻干燥,得PM-cc-DA固体,置于4℃下保存。用FT-IR及1H NMR对其结构进行表征,并通过其1H NMR图谱中PM的H1信号以及脱氧胆酸C18甲基氢信号进行取代度计算。
1.4 PM-cc-DA空白及载药胶束的制备
将20 mg PM-cc-DA聚合物溶解于20 m L PBS(p H 7.4,0.01 mol·L-1)中,充分混匀,于冰浴中超声以促溶,用0.45μm滤膜过滤除菌及其它不溶物,得1 mg·m L-1PM-cc-DA空白胶束溶液。
在避光条件下,将20 mg PM-cc-DOCA溶于20 m L甲酰胺溶液中,逐滴加入5 m L经过碱化的阿霉素溶液(1 mg·m L-1),充分溶解后,用水透析,用0.45μm滤膜以过滤除菌及其它不溶物,冻干,即获得PM-cc-DA/DOX载药胶束固体。
1.5 PM-cc-DA及PM-cc-DA/DOX表征
取2 m L新配制的胶束溶液,在25℃条件下用动态光散射仪测定其粒径分布、多分散系数(PDI)以及Zeta电势。用透射电镜(TEM)对其在溶液中的胶束状态进行表征,滴加PM-cc-DA空白胶束溶液于铜网,用2%磷钨酸染色5 min后充分干燥,在透射电镜下观察其胶束结构及形态。
1.6 PM-cc-DA聚合物临界胶束浓度测定
以芘试剂为荧光探针测定PM-cc-DA的临界胶束浓度。将PM-cc-DA溶于p H7.4的PBS中,配制1、0.5、0.1、0.05、0.01、0.001以 及0.000 1 mg·m L-1胶束溶液。将芘试剂溶于丙酮中配制成浓度为6.0×10-6mol·L-1的芘溶液,静置使丙酮挥发完全。将配制的PM-cc-DA胶束溶液加入离心管内中,充分混匀后静置过夜。设置激发光波长为334 nm,发射光波长为336及339 nm。以胶束浓度的对数值为横坐标,波长339及336 nm下的吸收强度之比为纵坐标作图,其切线交点处胶束浓度即为临界胶束浓度。
1.7 PM-cc-DA/DOX载药胶束载药性能测定
将5 mg盐酸阿霉素溶于5 m L体积比为1∶1的甲酰胺和水混合液中,配制成50μg·m L-1盐酸阿霉素溶液,用该溶剂将其继续稀释成不同浓度的盐酸阿霉素溶液,分别在480 nm下测定其吸光光度值,并绘制标准曲线,用于载药量及包封率的测定。
将2 mg PM-cc-DA载药胶束,用V(甲酰胺)∶V(水)=1∶1溶液混匀并定容至10 m L,测定其在480 nm下吸光度并计算其中阿霉素含量。根据标准曲线公式计算载药量x及包封率y(md:药物质量;mt:载药胶束质量;mu:加药质量)。
1.8 PM-ss-DA/DOX载药胶束氧化还原敏感性
将5 mg盐酸阿霉素溶于5 m L p H 7.4 PBS缓冲溶液中,配制成50μg·m L-1盐酸阿霉素溶液,用该溶剂将其继续稀释成不同浓度的盐酸阿霉素溶液,分别在480 nm下测定其吸光光度值,并绘制标准曲线,用于药物释放测定。
将PM-cc-DA/DOX载药胶束配置成1 mg·m L-1的胶束溶液,取2 m L加入到透析袋(MWCO 2000)中,置于50 m L p H 7.4的PBS缓冲溶液中,每隔一段时间取透析外液在480 nm波长下测定DOX的吸光度值,并通过标准曲线换算其浓度。同时对透析液补足等体积PBS缓冲溶液。每个时间点测量3个平行样。通过透析外液药物浓度绘制其对应药物释放曲线。
2 结果与讨论
2.1 DA-EDA的制备与结构表征
DA-EDA的制备是通过碳二亚胺试剂DIC进行活化后与经过氨基保护的己二胺中游离氨基反应生成酰胺键来获得,其反应式如图1所示。在DA活化过程中会产生大量N,N-二异丙基脲(DIU),而DIU的溶解度较低,可在反应体系中析出,因此,通过过滤方式除去DIU可降低产物中的杂质生成量。乙二胺的结构中有两个反应活性较强的氨基,它们极有可能都与DA反应而生成双取代乙二胺衍生物DA-EDA-DA,因此,需使乙二胺过量,并且采用滴加的方式,即,将活化产物滴加入乙二胺溶液中以降低DA-EDA-DA的生成。DA-EDA核磁谱图见图2。1H NMR:7.70(t,1 H,J=5.5 Hz),4.44(s,1 H),4.17(s,1 H),3.78(s,1H),3.37(tt,2 H,J=10.7,4.4 Hz,),3.01(q,2 H,J=6.3 Hz,),2.53(t,4 H,J=6.4 Hz,),0.92(d,3 H,J=6.5 Hz),0.84(s,3 H),0.59(s,3 H)。其 中3.01及2.53分 别 为C25及C26处亚甲基的质子信号,0.59,0.84,0.92分别为C-18、C-19、C-21处甲基的质子信号,说明乙二胺已成功连接到脱氧胆酸分子上。通过对C18位甲基(0.59)以及乙二胺中两个亚甲基(3.37,3.01)进行积分可知,该产物纯度接近99%。

图1 DA-EDA合成示意图Fig.1 Synthesis route of DA-EDA
2.2 PM-cc-DA的制备与结构表征
PM结构中含有大量亲水性羟基和羧基,水分子与它们形成强有力的氢键相互作用而很难被彻底除去,因此,在水相进行的EDC活化法更适合PMcc-DA的合成,其反应式如图3所示。由PM-cc-DA的1H NMR谱图(图4)可知,4.45、3.83、3.69、3.55分别归属为PM糖环上的H1、H2、H4、H3以及H5。3.16处为乙二胺连接臂中两个亚甲基质子信号,7.88为酰胺键质子信号。0.6~2.3之间的吸收峰为脱氧胆酸特征质子信号,说明DA-EDA已成功连接于PM中。对聚甘露糖醛酸H1位信号峰以及脱氧胆酸中C18位信号峰进行积分来对PM-cc-DA取代度进行计算,分别计算可得PM-cc-DA L取代度为7.9%,PM-cc-DA M取代度为16.7%,PM-cc-DA H由于其溶解度较差,无法通过1H NMR计算其取代度。

图3 PM-cc-DA合成过程Fig.3 Synthesis route of PM-cc-DA

图4 PM-cc-DA及PM的1 H NMR谱图Fig.4 1 H NMR spectra of PM-cc-DA and PM
2.3 PM-cc-DA空白及载药胶束的表征
PM-cc-DA空白胶束溶液的表观状态如图5所示。在PM-cc-DA 3种取代度的聚合物胶束溶液中,其取代度分别以L,M,H(低、中、高)代替,PM-cc-DA M的胶束溶液显示明显的蓝色乳光;而低取代度的胶束溶液无乳光,且不具有丁达尔现象;高取代度胶束溶解性较差,放置过程中即可出现聚沉现象,因此选用PM-cc-DA M(DS 16.7%)进行后续试验。

图5 PM-cc-DA空白及载药胶束溶液示意图Fig.5 Image of PM-cc-DA micelles and PM-cc-DA/DOX micelles
PM-cc-DA空白胶束溶液的粒径分布见表1。3批次不同PM-cc-DA胶束粒径呈现随取代度升高粒径变小的趋势,这是由于随着取代度升高,单个胶束分子中疏水嵌段增加,疏水端的吸引力逐渐增大,核壳界面的表面自由能逐渐变小,使得胶束趋于稳定;但是,取代度的过大,PM分子中的羧基减少,静电斥力相应减弱,从而导致胶束聚沉现象。表面电势也是纳米胶束的重要参数之一,直接影响其在介质中的稳定性以及整个体系的分散稳定性,其电势主要由PM中未被取代的羧基产生,这表明该胶束在体内可通过静电斥力获得抵抗粘附蛋白结合的能力,使其具有稳定性。

表1 PM-cc-DA空白胶束粒径分布表Table 1 Particle size distribution of PM-cc-DA micelles
将所制备胶束置于4℃冰箱内静置9个月后进行粒径测定,所测得对应粒径参数如表2所示。结果表明所制备胶束粒径均有一定程度的增大,PMcc-DA M胶束的平均粒径为(178.1±5.9)nm,多分散系数为(0.132±0.017),其粒径及PDI变化较少,证明其具有一定的稳定性。
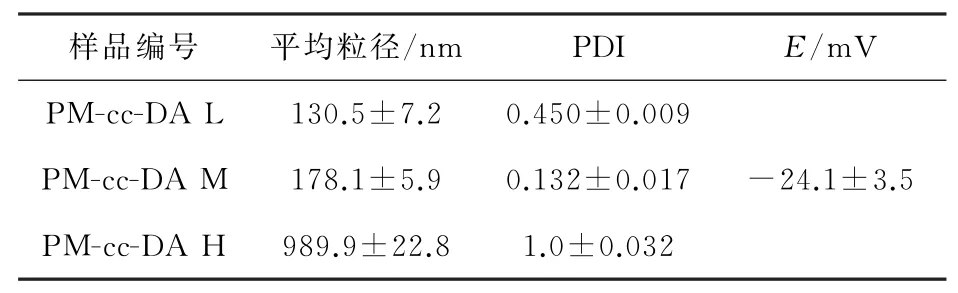
表2 静置9个月后PM-cc-DA空白胶束粒径分布表Table 2 Particle size distribution of PM-cc-DA micelles after 9 months
2.4 PM-cc-DA载药胶束的表征
通过透析法制备了PM-cc-DA的DOX载药胶束,其粒径参数如图6所示。结果表明,该胶束载药后平均粒径为186.9 nm,且粒径分布为0.183,说明其粒径分布较为集中。与PM-cc-DA空白胶束对比可知,该胶束载药后粒径有所增加,这与阿霉素的包载相关,阿霉素作为疏水药物,可通过疏水相互作用进入胶束的疏水内核,增大内核体积,并增强胶束的结构稳定性。PM-cc-DA/DOX载药胶束的载药量为13.7%,包封率为69.2%,对疏水性药物DOX具有良好的包载能力。

图6 PM-cc-DA载药胶束的粒径分布Fig.6 Particle size distribution of PM-cc-DA/DOX micelle
2.5 PM-cc-DA空白及载药胶束粒径透射电镜图像
通过TEM对PM-cc-DA及PM-cc-DA/DOX载药胶束进行表观状态表征,见图7。结果表明在透射电镜获得了两种胶束的负染图像,这是由于胶束内部为疏水结构,磷钨酸染色液无法进入胶束内部,使得胶束结构与周边背景颜色具有明显的衬度区别。TEM图像显示两种胶束结构完整,均具有良好的球状结构,并呈现较好的单分布状态。载药胶束与空白胶束相比粒径有所增大,这与药物与疏水内核结合使其增大相关。其粒径相比于动态光散射法所测得的粒径有所缩小,这与动态光散射法所测粒径是水合粒径有关,在制备透射电镜样品时需对所制备胶束进行干燥,这使得胶束与溶液中的状态有所差异,因此导致图像中粒径与所测粒径有所区别。

图7 PM-cc-DA空白及载药胶束TEM图像Fig.7 TEM images of PM-cc-DA and PM-cc-DA/DOX micelles
2.6 PM-cc-DA临界胶束浓度测定
通过芘试剂法对PM-cc-DA胶束的临界胶束浓度进行测定,由于芘试剂是一种对所在环境极性敏感的荧光制剂,因此可通过其荧光激发光谱中339 nm以及336 nm波长下的吸收强度比值来计算其临界胶束浓度,见图8。随着PM-ss-DA浓度的逐渐提升,胶束逐渐形成,芘试剂逐渐转移至胶束疏水内核,因此产生其荧光吸收峰的变化。通过计算芘试剂I339/I336与浓度的对数作图后所得的切线交点即可计算得到PM-cc-DA的临界胶束浓度,即为67.3 mg·L-1。这证明胶束可在较低的浓度下自发形成胶束,也意味着胶束具有在体液大量稀释下依旧保持胶束结构的稳定能力。

图8 PM-cc-DA胶束临界胶束浓度Fig.8 Critical micelle concentration of PM-cc-DA
2.7 PM-cc-DA/DOX药物释放曲线
通过在0.01 mol·L-1p H 7.4 PBS缓冲溶液中进行药物释放绘制了PM-cc-DA/DOX载药胶束的药物释放曲线,见图9。结果表明,药物在36 h下药物释放量为25.4%,这说明该胶束对于DOX具有良好的保护能力,在胶束通过EPR效应保留于肿瘤位置前能有效的将药物包覆在胶束内部,即,PMcc-DA/DOX载药胶束具有抗肿瘤药物的递送能力。

图9 PM-cc-DA/DOX载药胶束释放曲线Fig.9 Drug release profile of PM-cc-DA/DOX in PBS solution
3 结 论
以海洋天然多糖聚甘露糖醛酸以及疏水物质脱氧胆酸为原料制备了3批次两亲性聚合物PM-cc-DA,对制备的空白胶束PM-cc-DA的粒径及PDI参数进行对比选取PM-cc-DA M胶束进行载药试验,结果表明,该载药胶束载药量及包封率均较好,并且在体内p H环境下药物释放量较少,具有良好的抗肿瘤药物递送能力。
猜你喜欢
天然产物研究与开发(2022年1期)2022-11-27
中国临床医学影像杂志(2022年6期)2022-07-26
理化检验-化学分册(2021年10期)2021-11-29
湖北农业科学(2021年1期)2021-02-22
中国药学药品知识仓库(2021年17期)2021-01-11
消费导刊(2020年13期)2020-06-03
中国现代医生(2018年30期)2018-12-06
云南中医中药杂志(2016年12期)2017-03-04
创新时代(2016年4期)2016-05-17
药学与临床研究(2015年6期)2015-03-06
