
生发片中蒽醌类成分分析及其在不同生产工艺中的传递性
2022-01-27 10:44张亚莉谢诗婷苏薇薇姚宏亮
中成药 2022年1期
张亚莉, 刘 颖, 谢诗婷, 苏薇薇, 姚宏亮, 彭 维*
(1.广东省科学院动物研究所,广东省动物保护与资源利用重点实验室,广东省野生动物保护与利用公共实验室,广东 广州 510260;2.中山大学广东省中药上市后质量与药效再评价工程技术研究中心,广东 广州 510275;3.广西南宁百会药业集团有限公司,广西 南宁 530033)
生发片收载于《中药成方制剂》第十七册,由何首乌,女贞子,黑豆、黑枣、墨旱莲等12味中药组成,具有滋补肝肾,益气养血,生发乌发的功效,用于肝肾不足、气血亏虚所致的头发早白、脱落;斑秃,全秃,脂溢性脱发。近年来对生发片生产工艺、质量的研究较少,主要针对其中有效成分二苯乙烯苷和特女贞苷的含量[1]等。生发片处方中的君药用的是何首乌,具有一定肝毒性及泻下作用[2-6],致肝毒性的物质基础主要是其所含的蒽醌类成分大黄素及其衍生物、大黄酸[7-8];泻下作用的物质基础主要是结合蒽醌[9-10]。生发片服用疗程较长,为2周或1个月,故应建立蒽醌类成分检测方法。黑豆对何首乌有减毒的作用[11-12],生发片注册工艺中何首乌与黑豆、黑枣分开煎煮,厂家尝试何首乌与黑豆、黑枣合并煎煮,工艺流程见图1。本研究建立影响生发片安全性的蒽醌类成分总大黄素和结合蒽醌的检测方法,探讨不同煎煮工艺对两者的影响,并研究生产过程中其传递性规律,以期全面监控和评价该制剂质量和安全性。

图1 生发片工艺流程
1 材料
DMF-8A中药粉碎机(浙江温岭市铭大药材机械设备有限公司);MS205DU电子分析天平(十万分之一,瑞士梅特勒-托利多公司);HWS24型电热恒温水浴锅(上海一恒科技有限公司);KQ-250DE型数控超声波清洗器(昆山市超声仪器有限公司);Simplicity SIMS00000超纯水器(美国Millipore公司);UFLC超快速高效液相色谱仪(配置LC-20AD-XR二元泵、SIL-20AC-XR 自动进样器、CYO-20AC柱温箱、SPD-M20A PDA检测器,日本岛津公司);Triple TOF 5600+四级杆-飞行时间质谱仪、Sciex.Library数据库(美国AB SCIEX公司);Agilent 1260高效液相色谱仪(配置G1311B四元泵、G1316A柱温箱、G1329B进样器、G1315D DAD检测器);Ultimate 3000 DGLC高效液相色谱仪(配置DGP-3600SD双三元泵、SRD-3600脱气机、WPS-3000SL自动进样器、TCC3000-RS柱温箱、DAD检测器、Chromeleon7.2数据处理软件,美国Dionex公司)。大黄素(批号110756-201512,纯度98.7%)、大黄素甲醚(批号110758-201616,纯度99.0%)对照品(中国食品药品检定研究院)。甲醇、三氯甲烷(分析纯,广州化学试剂厂);盐酸(分析纯,成都市科隆化学品有限公司);甲醇、乙腈(色谱纯,美国霍尼韦尔公司);磷酸(色谱纯,上海阿拉丁生化科技股份有限公司)。生发片共11批,工艺研究用何首乌饮片、中间体及成品3批,均来源于广西南宁百会药业集团有限公司,具体见表1。
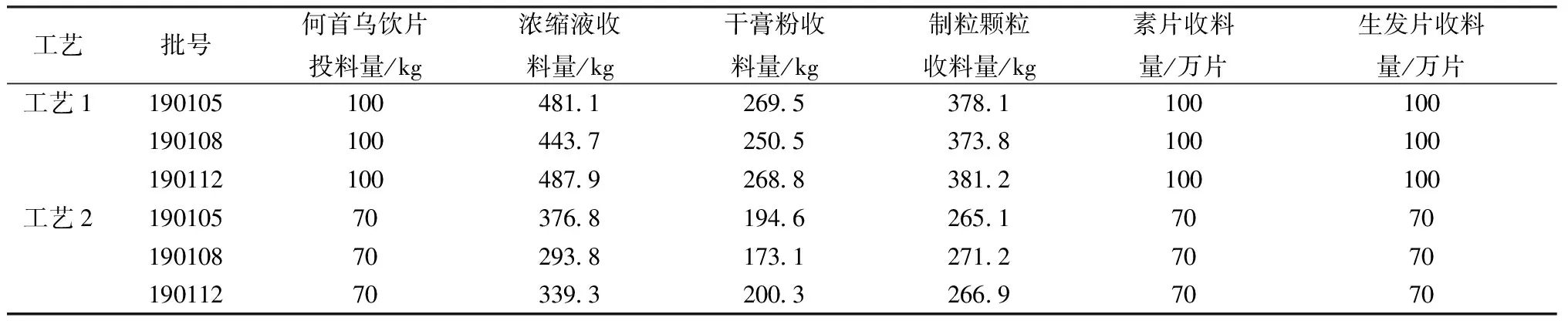
表1 生发片及其中间体信息
2 方法与结果
2.1 蒽醌类成分结构鉴定
2.1.1 供试品溶液制备 取本品适量,去糖衣,研细,取约0.5 g,精密称定,置于具塞锥形瓶中,精密加入10 mL甲醇,称定质量,超声(功率300 W、频率40 kHz)处理30 min,放冷,甲醇补足减失的质量,摇匀,滤过,取续滤液,即得。
2.1.2 色谱条件 Welch Ultimate XB-C18色谱柱(4.6 mm×250 mm,5 μm);流动相甲醇(A)-0.1%甲酸(B),梯度洗脱(0~105 min,5%~60%A;105~110 min,60%~90%A;110~115 min:90%~5% A);体积流量0.3 mL/min;柱温25 ℃;进样量10 μL。
2.1.3 质谱条件 ESI电喷雾离子源,电压-4 500 V;喷雾气3.8×105Pa;辅助加热气3.8×105Pa;离子源温度550 ℃;气帘气2.4×105Pa;碰撞气压力6.8×104Pa;扫描范围m/z100~2 000;负离子模式检测。
2.1.4 结果分析 总离子流图见图2,通过碎片离子分析及文献[13-14]报道,共鉴定了7种蒽醌类成分,见表2。

图2 生发片UFLC-Triple TOF-MS/MS总离子流图
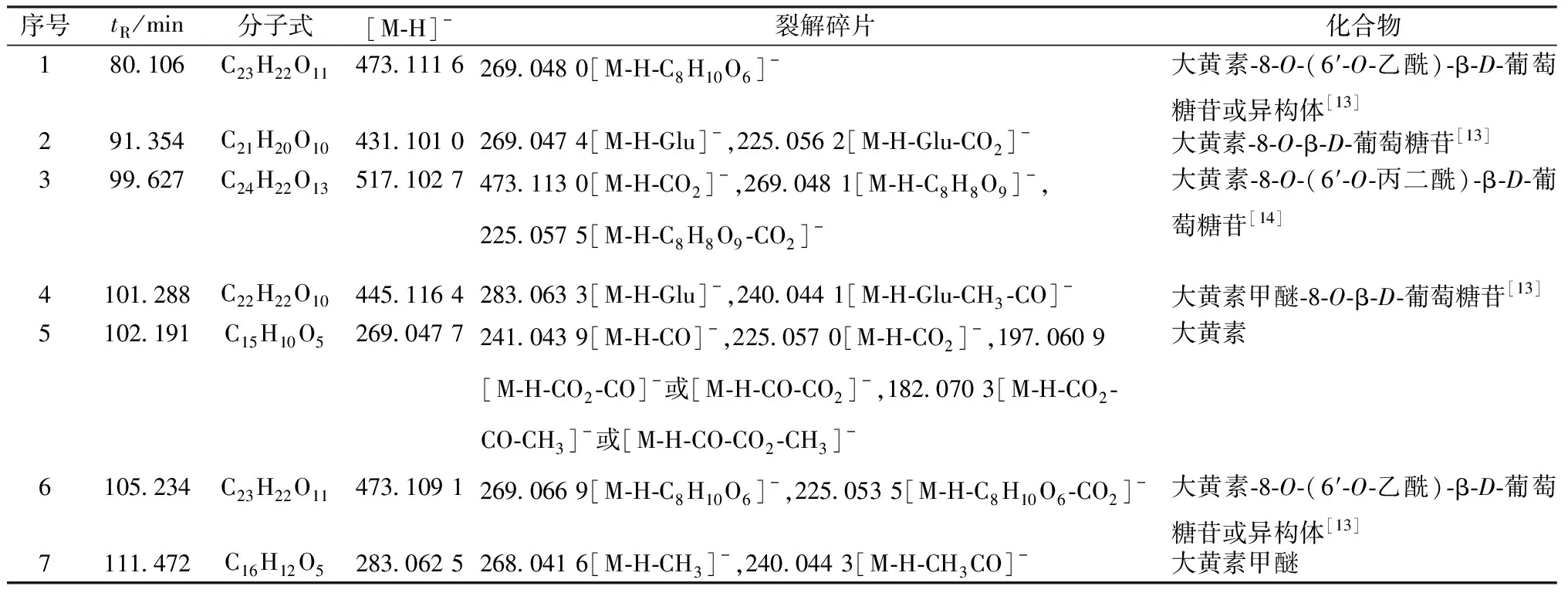
表2 蒽醌类成分鉴定结果
2.2 蒽醌类成分含量测定
2.2.1 对照品溶液制备 精密称取各对照品适量,甲醇制成分别含大黄素83.42 μg/mL、大黄素甲醚41.34 μg/mL的贮备液,各精密量取1 mL,置于10 mL量瓶中,甲醇定容至刻度,摇匀,即得(含大黄素8.342 μg/mL、大黄素甲醚4.134 μg/mL)。
2.2.2 供试品溶液制备 取本品适量,去除糖衣,研细,混匀,取约5.0 g(浓缩液5.0 g、干膏粉2.5 g、制粒颗粒5.0 g、素片5.0 g),精密称定,置于具塞锥形瓶中,精密加入50 mL甲醇,称定质量,超声处理30 min取出,放冷,甲醇补足减失的质量,摇匀,滤过,即得供试品溶液A(测定游离蒽醌用)。另取上述续滤液25 mL,置于具塞锥形瓶中,水浴蒸干,精密加入8%盐酸20 mL,超声(功率100 W、频率40 Hz)处理5 min,加三氯甲烷20 mL,水浴加热回流1 h,取出,立即冷却,置于分液漏斗中,少量三氯甲烷洗涤容器,洗液并入分液漏斗中,分取三氯甲烷液,酸液再用三氯甲烷振摇提取3次,每次15 mL,合并三氯甲烷液,回收溶剂至干,残渣加甲醇使溶解,转移至10 mL量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得供试品溶液B(测定总蒽醌用)。
2.2.3 阴性样品溶液制备 按照处方工艺,制备缺何首乌的阴性样品,按“2.3.2”项下方法制备,即得。
2.2.4 色谱条件 Welch Ultimate XB-C18色谱柱(4.6 mm×250 mm,5 μm);流动相甲醇(A)-乙腈(B)-0.1%磷酸(C),梯度洗脱(0~20 min,8%A,42%~50%B;20~30 min,8%A,50%B;30~38 min,8%A,50%~64%B;38~52 min,8%A,64%~68%B;52~55 min,8%A,68%B);体积流量1.0 mL/min;柱温25 ℃;检测波长254 nm;供试品溶液A、阴性样品溶液A进样量50 μL,对照品溶液、供试品溶液B、阴性样品溶液B进样量10 μL。
2.2.5 专属性试验 精密吸取对照品、供试品、阴性样品溶液适量,在“2.3.4”项色谱条件下进样测定,结果见图3~4。由此可知,蒽醌类成分分离度大于1.5,理论塔板数均不低于3 000,溶剂和阴性无干扰,表明该方法专属性良好。
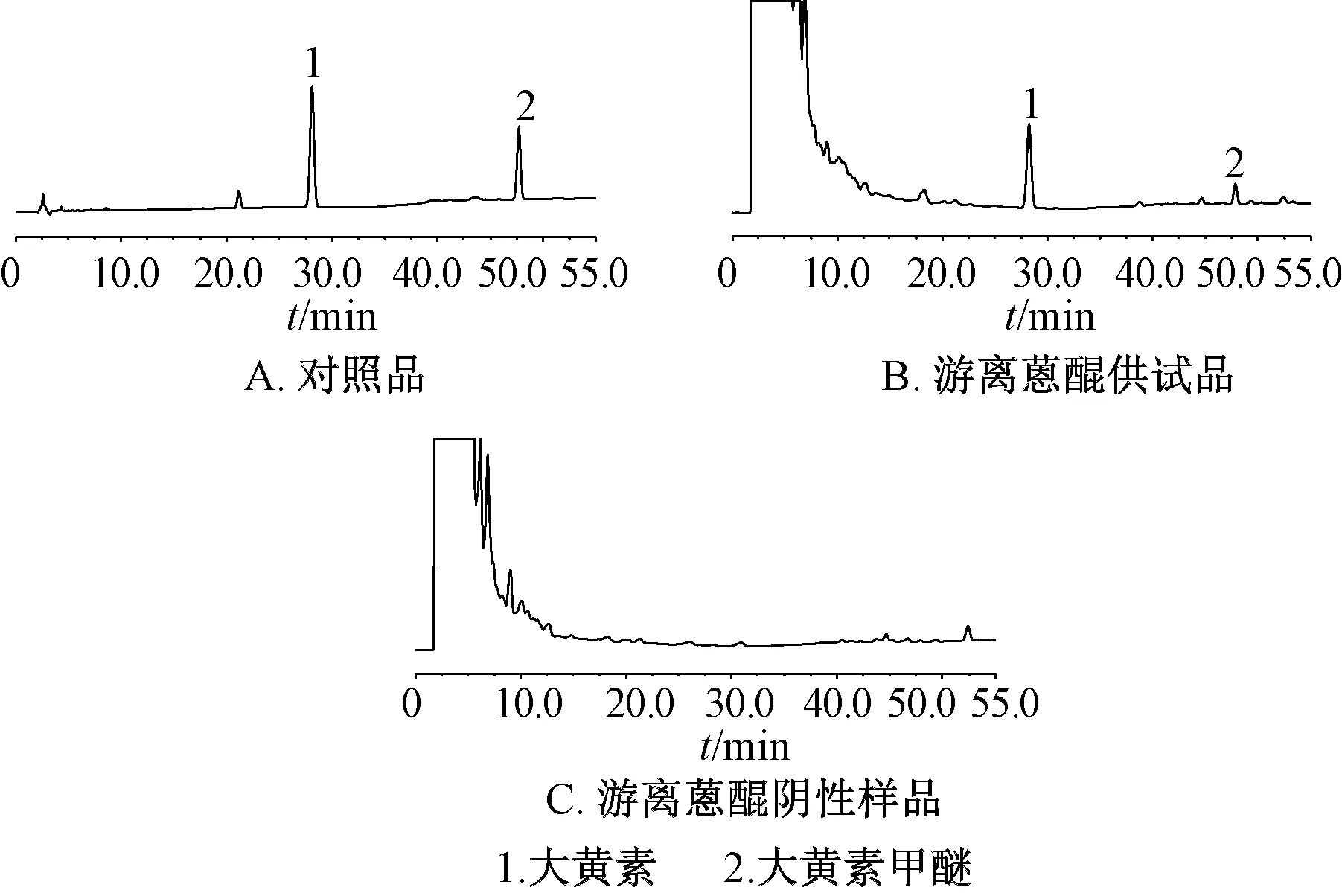
图3 游离蒽醌HPLC色谱图
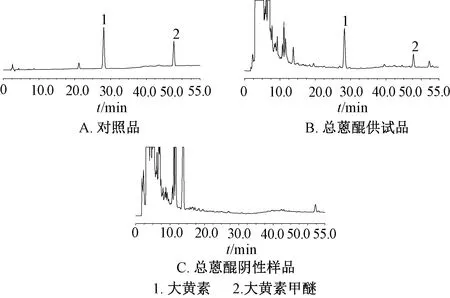
图4 总蒽醌HPLC色谱图
2.2.6 定量限 精密吸取“2.3.1”项下对照品溶液0.1 mL,置于10 mL量瓶中,甲醇稀释至刻度,在“2.3.4”项色谱条件下进样测定5次,测得大黄素、大黄素甲醚定量限分别为0.834 2、0.413 4 μg/mL,噪音比均大于10。
2.2.7 线性关系考察 精密吸取“2.3.1”项下对照品溶液0.1、0.5、1.0、1.5、2.0、2.5、3.0 mL,置于10 mL量瓶中,甲醇稀释至刻度,在“2.3.4”项色谱条件下进样测定。以对照品质量浓度为横坐标(X),峰面积为纵坐标(Y)进行回归,结果见表3,可知蒽醌类成分在各自范围内线性关系良好。
2.2.8 精密度试验 取“2.3.2”项下供试品溶液(批号1710062),在“2.3.4”项色谱条件下进样测定6次,测得游离大黄素、大黄素甲醚、总大黄素、大黄素甲醚峰面积RSD分别为0.38%、1.69%、0.27%、0.49%,表明仪器精密度良好。
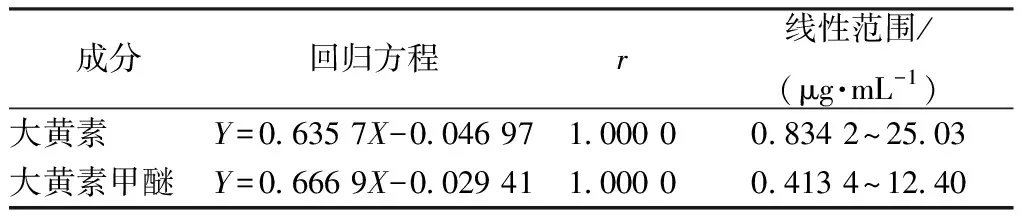
表3 蒽醌类成分线性关系
2.2.9 重复性试验 取同一批本品(批号1710062)适量,按“2.3.2”项下方法平行制备6份供试品溶液,在“2.3.4”项色谱条件下进样测定,测得游离大黄素、大黄素甲醚、总大黄素、大黄素甲醚含量RSD分别为2.81%、2.70%、3.06%、2.51%,表明该方法重复性良好。
2.2.10 稳定性试验 取同一份供试品溶液(批号1710062)适量,于0、2、4、8、12、24、48、72、120 h在“2.3.4”项色谱条件下进样测定,测得游离大黄素、大黄素甲醚、总大黄素、大黄素甲醚峰面积RSD分别为0.52%、0.92%、1.14%、1.41%,表明溶液在120 h内稳定性良好。
2.2.11 加样回收率试验 取蒽醌类成分含量已知的同一批(批号1710062)片剂,除去糖衣片,研细,分别取粉末约5.0 g(测定游离蒽醌用)、2.5 g(测定总蒽醌用),精密称定,各平行9份,置于具塞锥形瓶中,按照2015年版《中国药典》四部要求,使对照品加入量相当于样品量的50%、100%、150%,精密加入对照品溶液(含32.55 μg/mL大黄素、6.029 μg/mL大黄素甲醚,测定游离蒽醌用;含41.66 μg/mL大黄素、12.06 μg/mL大黄素甲醚,测定总蒽醌用)1、2、3 mL,各3份,按“2.3.2”项下方法制备供试品溶液,在“2.3.4”项色谱条件下进样测定,计算回收率。结果,游离大黄素、游离大黄素甲醚加样回收率分别为101.47%、100.98%,RSD分别为3.06%、3.72%;总大黄素、总大黄素甲醚加样回收率分别为107.35%、104.38%,RSD分别为3.20%、5.71%。
2.3 样品含量测定 取本品适量,按“2.3.2”项下方法平行制备2份供试品溶液,在“2.3.4”项色谱条件下进样测定,计算含量,结果见表4。
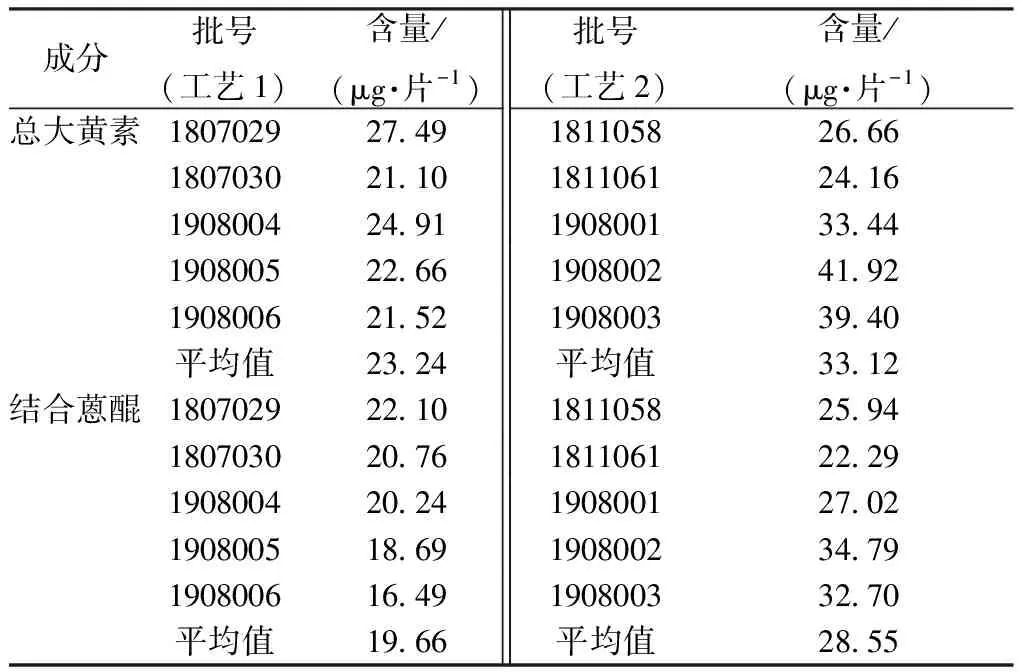
表4 蒽醌类成分含量测定结果(n=2)
2.4 传递性研究 结果见表5。

表5 饮片-中间体-成品蒽醌类成分的传递性测定结果(n=2)
3 讨论
3.1 安全性成分和限量的确定 生发片中蒽醌成分为游离蒽醌(大黄素、大黄素甲醚)和以大黄素、大黄素甲醚为母核的结合蒽醌,故本研究以总大黄素作为生发片肝毒性指标成分,结合型蒽醌(大黄素、大黄素甲醚)作为泻下指标成分。闵晓春等[15]曾以高剂量(12 mg/kg)大黄素灌胃给予大鼠连续2个月,发现无明显肝损伤,推算生发片中大黄素含量不超过7.41 mg/片可避免产生肝损伤风险。赵荣华[16]发现,何首乌高温清蒸后结合蒽醌含量在2.09 mg/g时(给药剂量25 g/kg)小鼠无泻下作用,推算生发片结合蒽醌含量小于66.99 mg/片时可避免发生腹泻风险。11批生发片蒽醌类成分含量远在安全限量以下,说明按疗程长期服用是安全的。
3.2 不同煎煮工艺分析 表5中生发片不同煎煮工艺饮片至浓缩液总大黄素、结合蒽醌转移率P值分别为0.09、0.18,均表明原注册煎煮工艺合理。
3.3 生产过程中蒽醌类成分传递规律分析 从表5转移率结果来看,煎煮浓缩、干燥环节是影响生发片安全性的关键步骤,制粒、压片到成品未受到高温影响的生产环节的蒽醌类稳定转移。
4 结论
本研究对生发片中蒽醌类成分进行了鉴定,同时建立了简便准确、专属性强的HPLC法测定其含量,证明了该制剂的安全性和生产工艺的合理性,揭示了蒽醌类成分的传递规律,为修订相关质量标准及控制其安全性、有效性提供了实验依据。
猜你喜欢
河南化工(2022年10期)2022-11-21
山西医科大学学报(2021年10期)2021-11-18
天津大学学报(自然科学与工程技术版)(2021年9期)2021-06-01
西北药学杂志(2021年1期)2021-02-03
世界科学技术-中医药现代化(2020年10期)2020-04-06
现代盐化工(2020年5期)2020-02-04
海峡姐妹(2019年8期)2019-09-03
家庭医药·快乐养生(2018年11期)2018-11-21
现代养生·上半月(2017年9期)2017-09-05
饮食科学(2016年4期)2016-07-06
