
SLC35A2基因新发突变致婴儿痉挛症1例并文献复习
2022-02-12 09:05林彬城
罕少疾病杂志 2022年2期
林彬城 陈 玲
福建医科大学附属厦门弘爱医院儿科 (福建 厦门 361006)
婴儿痉挛症(infantile spasms,IS)为一组常见的由多种病因导致的婴儿期癫痫综合征,是临床最常见的癫痫性脑病,据欧美统计发病率约为2.5~4.0/万人。1841年英国全科医生West[1]首次描述了这种独特的发作类型,所以又称West综合征。高峰起病年龄为4~8个月,仅有10%的患儿于1岁后起病[2]。主要特征是反复成簇的痉挛发作及发作间期脑电图呈高峰节律紊乱,常伴智力发育迟滞或倒退,45%的患儿合并孤独症谱系障碍[3]。IS发病机制复杂,病因多样,其中遗传性因素占重要地位。遗传学研究已经确定30多个基因中的致病变异是IS的病因,常见如STXBP1、GRIN2A、SCN2A、MAGI2、DEPDC5等,目前国内关于SLC35A2基因变异相关的报道较少。本文回顾性分析1例IS患儿的病例资料,辅助检查及遗传学检测结果,并复习相关文献,以期为该病的诊治提供依据。本研究获得厦门弘爱医院医学伦理委员会批准,患儿家长签署知情同意书。
1 病例资料
1.1 基本情况患儿女,4个月,因“反复抽搐2周”于2020年2月入住福建医科大学附属厦门弘爱医院。患儿于3月半龄时在安静状态下出现频繁抽搐发作,表现为双眼凝视、双上肢前举、头和躯干向前屈曲,呈点头拥抱样痉挛发作,成串发作,每串持续数分钟不等,约5~6串/天,无发热,无四肢强直,无二便失禁等,于厦门市妇幼保健院住院治疗(2020年4月28日至2020年5月1日)。予完善脑电图示:异常婴儿脑电图:(1)不典型高度失律;(2)监测到痉挛发作;诊断“婴儿痉挛症”,住院期间予静滴维生素B6(200mg q.d,4月28日至4月30日)、口服妥泰(3.125mg bid,4月28日至4月29日)、氯硝西泮(0.16mg qn,4月28日)等治疗,症状未明显改善,遂转诊厦门弘爱医院。患儿系第1胎第1产,足月顺产,出生体重3000g,出生后纯母乳喂养。患儿3个月会抬头,扶坐时头后仰,竖头稍不稳,双手不可主动抓握,追声追物可,对周围环境反应一般。
1.2 体格检查神志清楚,呼吸平稳,前囟平软,宽眉毛,宽鼻梁,厚嘴唇(图1A),竖头不稳,乳头内陷(图1B),心肺腹查体未见明显异常,四肢肌力、肌张力正常,拇指小,手指变细(图1C),脚趾重叠(图1D),病理征未引出。

图1A 宽眉毛,宽鼻梁,厚嘴唇;图1B 乳头内陷;图1C拇指小,手指变细;图1D 脚趾重叠。
1.3 辅助检查
1.3.1脑电图检查 不典型高度失律,监测到痉挛发作(图2)。

图2 脑电图
1.3.2 血尿代谢检查 丙氨酸氨基转移酶(ALT)62.5U/L,天门冬氨酸氨基转移酶(AST)129.0U/L,谷氨酰氨基转移酶(GGT)79.9U/L,血氨、血乳酸、血尿串联质谱未见明显异常。1.3.3 头颅磁共振 右侧海马前内缘可见一类圆形异常信号影,大小约5cm×6cm,考虑右侧海马脉络膜囊肿(图3)。
1.3.4 彩色多普勒超声检查 肝、胆、胰、脾未见明显异常,卵圆孔未闭状态,左室整体收缩功能正常。
1.3.5 遗传学检测 染色体核型分析未见异常;采用全外显子组测序方法。获得患儿监护人的知情同意后,采集患儿及其父母的外周血,委托北京赛福基因基于NovaSeq 6000技术测序平台,采用IDT xGen Exome Research Panel进行捕获建库,双末端(Paired-End)测序策略,明确IS致病基因,采用一代Sanger测序技术进行验证。基因检测发现患儿SLC35A2基因c.851T>C(p.Leu284Pro)位点的杂合突变,为疑似致病性变异,导致p.Leu284ProE氨基酸改变;其父母无此位点的突变,此突变为新发突变,见图4,为X连锁显性遗传。

图3A 头颅磁共振(矢状面);图3B 头颅磁共振(水平面,T2相)。
1.4 治疗情况予促皮质素(ACTH)12.5IU静滴(2020年5月8日至2020年5月12日),辅以口服托吡酯抗癫痫治疗,仍有痉挛发作2~4串/天,于5月13日改用ACTH 25IU静滴,5月21日发作停止,ACTH总疗程4周。
1.5 随访情况患儿出院后1.5个月未发作,于6.5月龄时再发痉挛,加用德巴金治疗,效果欠佳,于11个月龄时始减停德巴金,改用吡仑帕奈治疗,临床发作渐减少至停止。末次随访至1岁1月龄,目前能坐稳、扶站。
1.6 文献汇总以“先天性糖蛋白糖基化障碍Ⅱm型(早期婴儿型癫痫性脑病22型)”、“Congenital disorder of glycoprotein glycosylation Ⅱm(Early infantile epileptic encephalopathy type 22)”、“ SLC35A2”为关键词检索中国期刊全文数据库(CNKI)、万方数据知识服务平台、维普论文检索平台、生物医学文献数据库(PubMed)从建库至2021年1月收录的文献,排除重复报道的病例,共有基因确诊患者73例(国内7例,国外66例),其中男性9例、女性64例,病例资料相对完整的19例(表1~表3)。

表1 既往文献报道的先天性糖蛋白糖基化障碍Ⅱm型患儿及本例患儿的临床资料

表2 既往文献报道的先天性糖蛋白糖基化障碍Ⅱm型患儿及本例患儿的临床资料

表3 既往文献报道的先天性糖蛋白糖基化障碍Ⅱm型患儿及本例患儿的临床资料
2 讨 论
本研究共收集到临床数据相对完整的病例20例(其中既往文献19例,本文1例),以女性患儿居多(16/20,80.0%),男性患儿的临床特征与女性患儿不同,可能是因为保留了野生型SLC35A2等位基因[14];起病年龄多较小(≤6个月的占13/20,65.5%),大部分存在痉挛发作、发育迟缓(17/20,85.0%),可以合并小头畸形(8/20,40.0%)、面容异常(13/18,72.2%)、乳头内陷(5/10,50.0%)、骨骼异常(12/16,75.0%)、肌张力低下等(12/19,63.2%),部分还存在视觉异常(6/18,33.3%)、听力下降(3/11,27.3%)、头颅MRI多异常(16/20,80.0%)。脑电图多提示高峰节律紊乱(15/20,75.0%),超声心动图可有异常(5/12,41.2%),ACTH可能对这类患儿有效(7/14,50.0%)。
癫痫性脑病是指持续的癫痫活动本身可能导致严重的神经和认知障碍,婴儿痉挛症是最常见的婴儿癫痫性脑病,多在1岁内起病,4~8个月为高峰,主要临床特征为频繁的痉挛发作、脑电图特异性高峰失律、精神运动发育迟滞或倒退,其中发作间期的脑电图高度失律对本病有较大诊断价值。该病病因复杂,常见如遗传代谢病、神经皮肤综合征、围产期脑损伤等,而据统计,大约30%的病例仍然无法解释[15]。
对不明原因IS的个体的遗传研究已经确定了30多个基因的致病变异[16],其中有大部分都与其他癫痫类型有关。这些基因中的变异仅占未解决IS病例的11%~29%,说明尚有较大比例IS致病基因不明确(或为非遗传性)。既往报道和婴儿痉挛症相关的基因变异有SCN1A、KCNQ2、STXBP1、SCN2A、KCNB1、GNAO1、QARS和SLC35A2等。
基因检测作为诊断遗传性疾病的重要方法,已经在各类神经系统疾病的诊断、治疗中发挥了极大的作用。S的基因突变类型包括错义突变、无义突变等各种类型,以错义突变最常见[17]。
溶质载体家族35成员A2基因(SLC35A2基因)是溶质载体家族35基因中的一员,(OMIM:314375),编码尿苷二磷酸半乳糖转运体,位于染色体Xp11.23,(NM_001042498.3)包含4个外显子,编码393个氨基酸,负责将半乳糖从胞质运输到高尔基体或内质网中[18]。SLC35A2基因变异导致半乳糖不能进入高尔基体,产生缺乏半乳糖的截短聚糖,引起糖蛋白、糖脂和蛋白多糖的半乳糖化降低,导致先天性糖基化障碍 (congenital disorders of glycosylation,CDG)Ⅱ型,即早期婴儿型癫痫性脑病22型,是一种X连锁显性遗传的严重神经系统疾病,以婴儿期癫痫发作、发作间期脑电图高度失律、 肌张力减退和严重的智力残疾伴语言缺乏为特征。颅脑畸形包括大脑和小脑萎缩,胼胝体薄也很常见,且所有患者的胼胝体都比年龄正常值短[17]。此外,部分患儿有畸形特征或粗糙面容。
糖基化是将复杂的糖链(聚糖)添加到蛋白质和脂质的过程。蛋白质糖基化包括内质网(ER) -高尔基体网络中的N-链和O-链糖基化途径[11]。糖基化过程的缺陷导致先天性糖基化疾病(CDG),是一个不断扩大的群体[19]。迄今为止,已知的由不同糖基化途径缺陷导致的罕见人类遗传疾病达100多种,其中超过80%与神经异常有关[17],常表现为结构异常、发育迟缓及癫痫发作。这些缺陷中的大多数最初可以通过碳水化合物缺乏转铁蛋白(CDT)分析,检查血清中丰富的转铁蛋白(Tf)糖基化状态[19]。转铁蛋白质谱与常规筛选试验相比,在识别SLC35A2-CDG方面更具有特异性。
关于SLC35A2-CDG的30例回顾性研究[19]发现,患儿有不同程度的躯体和神经发育缺陷(图5)。其中,发育迟缓(30/30,100%),癫痫(28/30,93%);小头畸形(13/30,43%);面容异常(26/30,87%);皮肤异常包括乳头内陷(8/30,27%)、异常色素沉着(4/30,13%);骨骼异常(25/30,83%),通常表现为多重特征,包括四肢短小(9/30,30%)、挛缩(8/30,27%)、脊柱侧凸(15/30,50%)和手或手指畸形(16/30 ,53%), 这些患儿可能是因为更严重的蛋白多糖合成障碍;脑影像异常(25/30,83%),其中蛋白质异常(16/30,53%)、小脑萎缩(17/30,57%)。与本研究所述基本一致,提示SLC35A2-CDG典型的临床特征包括癫痫发作、发育迟缓、面容粗糙、骨骼异常、肌张力低下,乳头内陷等。Vals等人在同一个研究中[19]描述了另外15例具有相同突变的患者,80%的患儿存在癫痫,9例患儿存在心律失常或与未确定的癫痫性脑病相一致的表现。基于以上信息,SLC35A2基因被假设为癫痫性脑病的候选基因。另文献报道2例患儿在婴儿期行头颅MRI检查正常,后期随访复查时出现异常,提示SLC35A2基因变异相关的脑结构异常可能随着时间的推移而演变,建议定期随访头颅影像学检查[10]。Quelhas等[12]的研究中该患儿无癫痫且伴有血清IGF1降低、轻度神经系统受累、轻度面部畸形和手指、脚趾畸形,提示SLC35A2-CDG也有不典型的轻型病例。
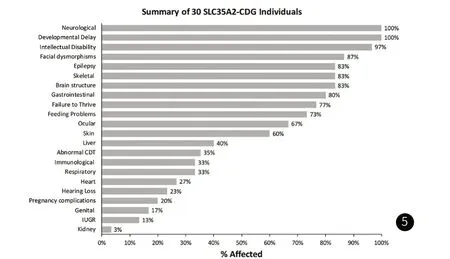
图5 图例:关于SLC35A2相关糖基化障碍的30例回顾性研究4.28-4.30是日期,就是指2021.04.28-04.30
研究报道,补充半乳糖几乎可以完全修复由于SLC35A2突变所致的糖基化障碍[20]。田杨等[5]报道的SLC35A2基因突变致West综合征的患儿给予食品添加剂来源的半乳糖(纯度80%),每天1g/kg,1个月后患儿大运动和精神反应显著改善,可以快速地翻身和响亮地发声,癫痫发作明显减少,且痉挛动作幅度减轻,远期疗效有待继续观察。Witters等[21]关于10例SLC35A2-CDG患儿在补充半乳糖18周后,5名患儿恢复发育进展,包括姿势控制、对刺激的反应、咀嚼和吞咽的改善。此外,胃肠道症状和癫痫也有所改善。总体来看,口服半乳糖耐受性好,有望成为SLC35A2-CDG的饮食疗法,但作为一种较新颖的治疗方法,应用前应获得家属的充分知情同意。
吡仑帕奈(perampanel)是第3代的新型的抗癫痫发作药物,为α-氨基-3-羟基-5-甲基-4-异恶唑丙酸(AMPA)受体拮抗剂,通过AMPA受体选择性和非竞争性拮抗作用,直接减弱谷氨酸引起的神经过度兴奋而抑制癫痫发作[22]。根据一项先前的报告[23]评估吡仑帕奈的耐受性和有效性在24例难治性癫痫患儿中,42%的患儿有癫痫发作减少50%或更多,8%的患儿癫痫发作减少33%,4%的患儿癫痫发作不那么严重。本例患儿初始应用ACTH及口服多种抗癫痫发作药物后效果欠佳,予改用吡仑帕奈后目前控制尚可,有待进一步随访观察疗效。
综上,本文致力于探讨SLC35A2、CDG和IS之间的关系,进一步描述与SLC35A2致病变异相关的表型。证据已表明该基因变异可导致相对典型的临床症状、体征,如痉挛发作、发育落后,提示SLC35A2基因可能是婴儿痉挛症的致病基因,且有一定的基因型-表型相关性,有待进一步的功能验证研究。然而该变异的作用机制目前尚未明确。尽管如此,临床医生遇到患有早发性癫痫性脑病伴发育落后,且糖基化检测异常的患儿,应从遗传学角度考虑疾病,检测如SLC35A2基因以及CDG以明确病因,从而提高明确IS病因的能力。基因检测是最有潜力的一种罕见病诊断方法,不但能验证疾病的临床诊断、提供遗传咨询服务,还可以用于致病基因携带者的检测[24-25]。目前SLC35A2基因变异相关CDG尚无特效治疗,补充半乳糖可作为一种尝试性治疗。对于不同病因导致的婴儿痉挛症,ACTH的疗效并不一致,对于ACTH应答欠佳的病例,新型抗癫痫药吡仑帕奈可能是有潜质的治疗选择。
猜你喜欢
安徽农业大学学报(2022年2期)2022-11-09
中国生物化学与分子生物学报(2022年8期)2022-09-08
中华实用诊断与治疗杂志(2022年1期)2022-08-31
祝您健康(2022年2期)2022-01-14
家庭医药(2020年10期)2020-10-30
三农资讯半月报(2020年8期)2020-05-13
北京广播电视报(2019年10期)2019-04-22
家庭医学(2017年12期)2018-01-15
家庭医学(2014年6期)2014-09-11
浙江中医杂志(2004年2期)2004-09-14
