
铁原子吸附联苯烯单层电子结构的第一性原理*
2022-02-17 02:25吴洪芬冯盼君张烁刘大鹏高淼闫循旺
物理学报 2022年3期
吴洪芬 冯盼君 张烁 刘大鹏 高淼 闫循旺†
1)(曲阜师范大学物理工程学院,曲阜 273165)
2)(宁波大学物理科学与技术学院物理系,宁波 315211)
联苯烯单层由碳原子的四元、六元和八元环组成,具有与石墨烯相似的单原子层结构.2021 年5 月,Science 首次报道了该材料的实验合成,引起了科研工作者的极大关注.基于第一性原理的密度泛函方法,研究了铁原子在联苯烯单层的吸附构型并分析了其电子结构.结构优化、吸附能和分子动力学的计算表明,联苯烯单层的四元环空位是铁原子最稳定的吸附位点,吸附能可达1.56 eV.电子态密度计算表明铁3d 电子与碳的2p 电子有较强的轨道杂化,同时电荷转移计算显示铁原子向近邻碳原子转移的电荷约为0.73 个电子,说明联苯烯单层与吸附的铁原子之间形成了稳定的化学键.另外,铁原子吸附于联苯烯单层后体系显磁性,铁原子上局域磁矩大小约为 1.81 μB,方向指向面外.因此,本文确认了联苯烯单层是比石墨烯更好的铁原子吸附载体且体系有磁性,这为研究吸附材料的电磁、输运、催化等特性提供了新的平台.
1 引言
石墨烯由于其奇特的二维蜂窝状结构以及在电、磁、热、力学等方面独特的物理化学性质,引起了科研工作者极大的兴趣.自2004 年石墨烯被制备以来[1],探索新型的低维材料便一直是凝聚态物理领域的前沿课题,其中二维的碳材料是该领域科研工作者的重点研究对象之一[2-7].近年来,虽然理论上已经预测了20 余种[8-31]碳的二维网格结构,如石墨炔(graphyne)[8,9]、石墨双炔 (graphdiyne)[10,11]、石墨n 炔(graphenylene)[12,13]、联苯烯单层(biphenylene)[14,15]、轴烯(radialenes)[16]等,但遗憾的是,到目前为止,实验上所合成的二维碳材料非常少,主要有石墨烯[1]、石墨双炔[11]、γ-石墨炔(γ-graphyne)[32]、tetrakis-DBAs[33]等.
近日Fan 等[2]在实验上成功制备了石墨烯二维同素异形体—联苯烯单层并发表在Science上,引起相关科研工作者的广泛关注.该材料最早由Balaban 等[34]于1968 年理论预测得到.联苯烯单层有类石墨烯的二维单层结构,二者均由碳元素组成,每个原子都是三配位的碳原子,其稳定性都来源于sp2杂化轨道的成键机制.石墨烯是最典型的二维材料,也是人们研究最广泛的二维材料,具有独特的晶体结构和电子结构,从而具备优异的力学、热学、电学性质,在能源、传感器、半导体、生物医学等多个领域有广泛的应用潜力.鉴于联苯烯单层与石墨烯的相似性,联苯烯自身的研究具有重要的科学意义.另外,石墨烯的化学修饰进一步丰富了石墨烯基材料的功能,如可作为电极材料、储能材料、催化材料、自旋电子器件材料等.相比之下,联苯烯的化学修饰会带来更多更复杂的物理特性.这是因为联苯烯单层由四元环、六元环和八元碳环组成,而石墨烯中只有六元环,故联苯烯可以提供更多样化的化学修饰环境.因此,联苯烯及化学修饰的联苯烯材料有什么样的奇异物性很值得期待,另一方面确定其基本电子结构也是当前的一个紧迫任务.
铁原子作为一种极具代表性的过渡金属原子,吸附或嵌入在二维单层中形成复合材料,在自旋电子器件[35]、催化[36]、能源(电池)等诸多领域有着巨大的研究价值和广泛的应用前景.以石墨烯为代表的二维碳材料吸附过渡金属原子后常表现出有趣的磁性质[37];此外,碳基铁原子催化剂是氧还原等反应中活性最高的催化剂之一[38],由于极高的原子利用率,明确的活性位点和长期稳定性等特点,其具有优异的催化性能与巨大的应用前景[39,40].而联苯烯单层作为一种新兴的二维碳材料,其吸附铁原子的性质尚未被研究.
本文采用基于第一性原理的密度泛函理论方法,研究了铁原子在联苯烯单层的吸附构型并分析了吸附前后的电子结构.结果表明,联苯烯单层是铁原子的良好载体.对于铁原子的吸附,联苯烯单层有着比石墨烯更大的吸附能,二者之间存在明显的电荷转移和稳定的化学键.铁原子的吸附使联苯烯单层变为磁性金属材料.这对小型化自旋电子器件及单原子催化剂等领域的研究具有参考价值.
2 计算方法
本文计算使用基于第一性原理的VASP 软件包(Viennaab-initosimulation package)[41,42].采用投影缀加平面波方法[43]赝势来描述离子实与电子的相互作用,交换关联泛函采用Perdew-Burke-Ernzerhof 形式[44]的广义梯度近似(generalized gradient approximation,GGA).采用GGA+U方法和VDW-D3 方法考虑了d 电子态在位库仑能修正和范德瓦耳斯作用[45,46].结构优化和自洽计算的平面波基组展开的截断能设置为500 eV,结构优化的总能量和力的收敛阈值分别为10—5eV 和0.01 eV/Å.进行自洽计算时采用了18×15×1的k点网格,确保k点间隔在倒空间中小于 2π×0.005 Å—1.联苯烯单层位于晶胞的xy平面,采用联苯烯单层的 3×3×1 超胞构建吸附模型,在z方向上取18 Å的真空层以模拟分离的联苯烯单层.用从头算分子动力学(ab-initiomolecular dynamics,AIMD)模拟来检验热稳定性.采用粒子数-体积-温度(NVT)系综,温度保持在1000 K,持续5 ps,时间步长为1 fs[47].
3 结果与讨论
3.1 吸附模型
如图1(a)所示,根据对称性,石墨烯有3 个可能的原子吸附位点,分别是顶位(T)、空位(H)和桥位(B).结果表明,铁原子可以稳定吸附于空位(H)上,吸附高度为1.53 Å,铁-碳键长为 2.11 Å,这与已有的结果相吻合[48],说明本文中计算参数的选取是合理的;联苯烯单层是一种由四元、六元和八元的碳环组成的单原子层二维材料,图1(b)的黑色虚线框展示了该材料的最简原胞.由于对称性的降低,联苯烯单层需要考虑更多的吸附位点,分别是2 个顶位(T1,T2)、3 个空位(H1,H2,H3)和4 个桥位(B1,B2,B3,B4),在图1(b)中分别由绿、红、蓝点标注.图1(c)—(h)展示了铁原子在H1,H2,和H3位点的吸附模型,经过完全的结构弛豫计算发现,铁原子可以稳定地吸附在联苯烯单层的H1,H2和H3位点.在H3位点吸附时,铁原子倾向于吸附在八元环空位的一侧,且会导致联苯烯单层发生明显的晶格畸变.

图1 (a)石墨烯和(b)联苯烯单层及其需考虑的吸附位点;(c),(f);(d),(g);(e),(h)分别是铁原子吸附于联苯烯单层四、六和八元环空位(H 位点)的俯视图及侧视图Fig.1.(a)and (b)Graphene and biphenene networks and the adsorption sites of Fe atoms;(c)and (f),(d)and (g),(e)and (h)are top and side views of Fe atoms on the top of 4-,6-,and 8-membered ring (H sites)of biphenene networks,respectively.
3.2 吸附能
为检查吸附的稳定性,首先计算吸附能,计算公式如下:

式中,Etot表示载体吸附原子后的总能量;Eslab表示未作吸附时的原始联苯烯单层的能量;EFe表示铁的自由原子的能量,越大的吸附能,意味着铁原子在能量上越利于吸附.如表1 所列,计算铁原子在石墨烯和联苯烯单层上的吸附能时发现,对石墨烯而言,铁原子在其空位(H)的吸附能,明显大于在顶位(T)和桥位(B)的吸附能,即从能量角度看,石墨烯的空位是铁原子的最稳定吸附位点;对于联苯烯单层来说,铁原子在其四元环和六元环空位 (H1,H2和H3)的吸附能明显较大,H1,H2和H3位点都是铁原子的稳定吸附位点,其中四元环空位的吸附能最大,是铁原子的最稳定吸附位点;联苯烯单层的顶位(T1,T2)和桥位 (B1,B2,B3,B4)的吸附能虽都为正值,但结构优化表明这些吸附构型并不能稳定存在,而且,与H1和H2位点相比,吸附能明显较小,即从能量角度看铁原子并不倾向于在这些位点形成稳定的吸附.

表1 铁原子吸附在石墨烯和联苯烯单层各位点的吸附能Table 1. Adsorption energy of Fe atom adsorbed on each point of graphene and biphenene.
石墨烯H 位点对铁原子的吸附能为0.84 eV,由于吸附能较低,在实际应用中石墨烯难以作为铁原子的载体.铁原子在联苯单层 H1和H2位点的吸附能大小分别为1.56 和1.53 eV,几乎是铁原子在石墨烯上吸附能的2 倍.从能量角度看,联苯烯单层对铁原子的吸附比石墨烯更为稳定,说明联苯烯单层是过渡金属原子的理想吸附载体,可作为电、磁、热、催化等功能材料的理想平台.
3.3 分子动力学
通过第一性原理的AIMD,进一步检查了联苯烯单层空位(H1,H2和H3)吸附铁原子的热力学稳定性.在1000 K 下,铁原子吸附于联苯烯单层的四元、六元和八元环后能量随时间的演化曲线见图2,其中还给出了对应1000 K 下经过5 ps 后的吸附情况的俯视图和侧视图.如图2(a)和图2(b)所示,当铁原子吸附于四元环和六元环空位时,能量随时间的变化曲线在一定的区间内波动,且不会出现明显的能量下降,吸附铁原子后的联苯烯单层的二维结构能在1000 K 下很好地保持,铁原子能够稳定地保持在四、六元环的空位(H1,H2位点).如图2(c)所示,当铁原子吸附于联苯烯八元环空位时,能量曲线随时间的变化曲线在一定区间内,有着相对较大的起伏,由于八元环有着较大的空间,在热扰动下,铁原子会在八元环内作往复运动,运动过程如图2(d)中的①,②,③,④和⑤所示.该吸附特点是石墨烯等材料所不具备的.

图2 (a),(b)和(c)是在1000 K 下,铁原子分别吸附于联苯烯单层的四、六和八元环后能量随时间的演化曲线;(d)在1000 K 下,铁原子在八元环空位的运动情况Fig.2.(a),(b)and (c)Time evolution curves of the energy of Fe atoms adsorbed on the 4-,6-and 8-membered rings of the biphenene network at 1000 K,respectively;(d)motion of iron atom absorbed on hollow site of 8-membered ring at 1000 K.
3.4 电荷转移
3.4.1 Bader 电荷
利用Bader 电荷分析[49],定量计算了铁原子与联苯烯单层(H1,H2和H3)间的电荷转移情况.当吸附在石墨烯空位时,每个铁原子失去0.80 e,最近邻和次近邻碳原子得电子,每个最近邻碳原子得电子约为0.10 e.与石墨烯的吸附情况类似,当铁原子吸附在联苯烯单层时,电荷从铁原子转移至近邻碳原子上.在四元环空位吸附时,铁原子失去0.73 e,每个最近邻的碳原子得电子为0.14 e,其余电子转移到次近邻碳原子上;联苯烯单层中的六元环为压扁的六元环,从铁原子看,有四个最近邻和两个次近邻碳原子,铁原子失去0.83 e,每个最近邻碳原子得0.23 e,另外两个次近邻碳原子各失去0.06 e;在八元环空位时,铁原子上失去0.69 e,每个最近邻碳原子得到0.15 e.这些电荷转移说明铁原子与联苯烯单层有着紧密的结合,与石墨烯情况相比,最近邻碳原子上获得的电荷数增多,所以有着更大的吸附能.
3.4.2 差分电荷密度分析
通过差分电荷计算,进一步分析了铁原子在联苯烯单层上的电荷转移情况,计算方法如下:

式中,ρtot表示联苯烯单层吸附铁原子后的总电荷密度,ρslab表示未作吸附时的原始载体的电荷密度,ρFe表示孤立铁原子的电荷密度.
如图3 所示,黄色部分表示电荷密度增加,蓝色表示电荷密度减少.由图3 中黄色区域可以看出,铁原子和联苯烯单层间出现了电荷的聚集,这与Bader 电荷分析中铁原子对联苯烯单层转移电荷的结果相一致,表明联苯烯单层与铁原子之间形成了稳定的化学键.
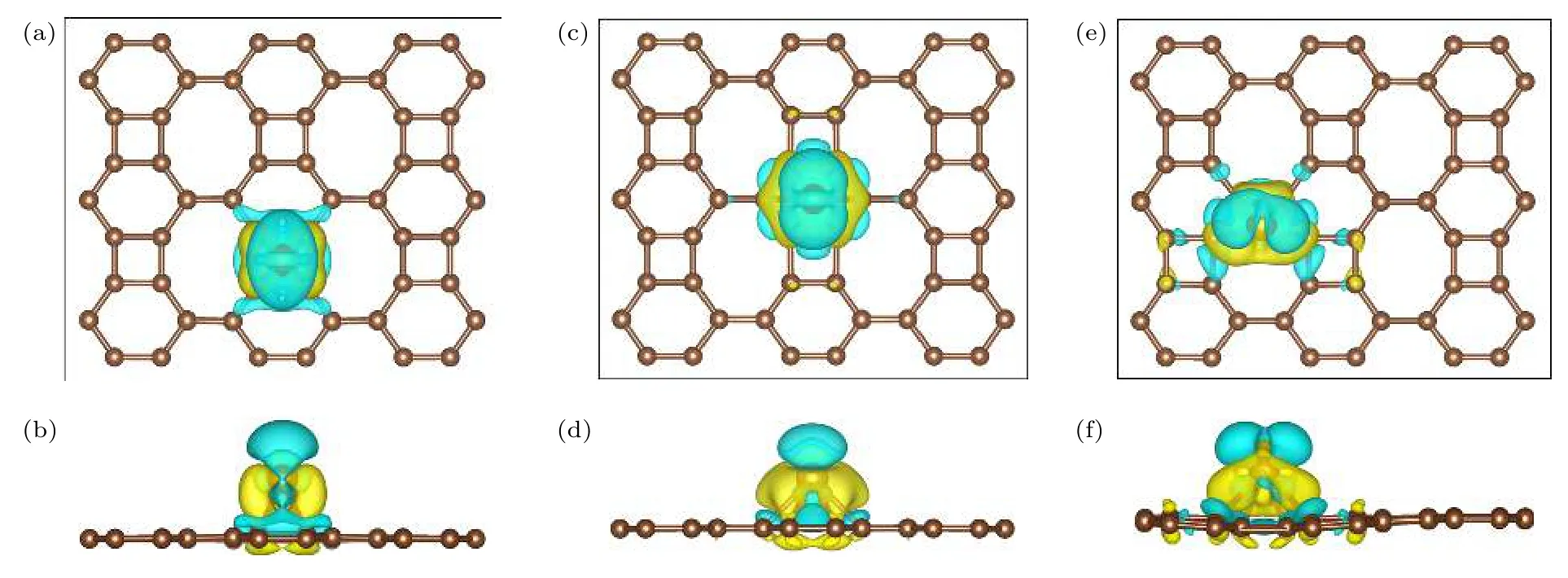
图3 (a),(b);(c),(d);(e),(f)分别是铁原子吸附于联苯烯单层四、六、八元环空位(H 位点)情况下的差分电荷俯视图及侧视图Fig.3.(a),(b);(c),(d);(e),(f)are top and side views of differential charge when Fe atoms are adsorbed on top of 4-,6-and 8-membered ring (H sites)of biphenene networks,respectively.
3.5 电子结构
3.5.1 石墨烯和联苯烯单层
石墨烯与联苯烯单层在—4.0—3.0 eV 能量范围内的能带与电子态密度如图4(a)和图4(b)所示.由费米能级附近的电子态分布可见,联苯烯单层与石墨烯有着截然不同的电子性质,石墨烯是零能隙的半导体,费米能级处的电子态密度为零,在倒空间的K点存在“狄拉克锥”;而联苯烯单层有两条能带穿过费米能级,不再具有“狄拉克锥”的能带特征,在费米能级处态密度有一定的数值,呈现金属性.对联苯烯单层来说,不用掺杂就有一定的载流子浓度,可作为导电薄层或条带应用于量子器件.

图4 (a)石墨烯和(b)联苯烯单层的能带及态密度图(费米能级设为0 eV)Fig.4.Energy band and density of states of (a)graphene and (b)biphenene network.The Fermi energy is set to zero.
3.5.2 铁原子吸附于联苯烯单层四元环空位
以铁原子的最稳定吸附位点为代表,图5(a)展示了当铁吸附于联苯烯单层的四元环空位时,铁的3d 轨道及其最近邻碳原子2p 轨道的投影态密度.在费米能级以下,铁的3d 轨道和碳的2p 轨道有较强的杂化,结合Bader 电荷中吸附的Fe 原子向近邻碳原子转移的电荷约为 0.73 e,进一步说明铁原子与联苯烯单层之间能够形成较强的相互作用.在费米能级附近 (—3.0—2.0 eV),体系的电子态主要由铁的3d 轨道主导.同时,铁3d 电子态有较强的自旋极化,从而形成局域磁矩,使铁原子与联苯烯单层的复合体系表现出明显的磁性.此外,在—0.23 eV 处,自旋向下的电子态存在着极强的态密度尖峰.图5(c)显示了铁3d 轨道的分轨道dxy,dyz,dxz,,由图5(c)可以看出,该尖锐的态密度峰来源于铁的轨道.这是由于垂直于联苯烯单层的轨道难以与近邻碳原子的p 轨道杂化造成的,与体系的准二维特性密切相关.

图5 铁原子吸附于联苯烯单层四元环空位时的投影态密度图(费米能级为零)(a),(c)GGA 计算;(b),(d)GGA+U 计算Fig.5.Projected density of states of Fe atoms adsorbed on the top of 4-membered ring of biphenene network:(a)and (c)GGA calculations;(b)and (d)GGA+U calculations.The Fermi energy is set to zero.
自旋极化计算表明,吸附铁原子后的联苯烯单层为磁性金属材料.当铁原子吸附于联苯烯单层四元环空位时,磁矩大小约为 1.81μB.进一步检查材料的非共线磁性发现,当铁原子吸附于联苯烯单层四元环空位时,易磁化轴指向面外,铁原子的磁各向异性能大小为0.64 meV,与常见的二维磁性材料 CrXTe3和Fe2Si 相当[50,51].
3.6 磁 序
石墨烯在比表面积、机械强度、电子迁移速度、热导率等方面有着明显的优势,但石墨烯本身是非磁性的,限制了石墨烯在自旋电子器件方面的应用.自石墨烯发现以来,研究人员采用缺陷、吸附、掺杂、边界等方式实现石墨烯基复合材料的磁性[52].由于石墨烯的吸附位点(六元环空位)太过密集且吸附能较小,不利于形成规则的磁原子阵列,又因为联苯烯单层的四元环空位形成分布稀疏、间距合理的吸附位点网格,且吸附牢固,所以,以联苯烯为基底,四元环空位吸附上铁原子,易于形成稳定的磁有序结构.这是联苯烯相对于石墨烯一个明显的优点.本研究小组设计了几种磁序,如图6 所示,分别命名为铁磁(FM)、共线反铁磁序一(Coll-I)、共线反铁磁序二(Coll-II)和奈尔反铁磁序(Nèel),用标准GGA 方法分别计算了相应的能量,并显示在表2 第1 行中.这些数据显示奈尔反铁磁序最稳定,从而确定均匀吸附铁原子的联苯烯单层具有反铁磁性.

表2 铁原子吸附联苯烯单层不同磁序下的能量Table 2. Energies of Fe atom adsorbed biphenene layer in various magnetic orders.

图6 联苯烯单层均匀吸附铁原子后的几种磁序结构(a)铁磁(FM);(b)共线反铁磁序一(Coll-I);(c)共线反铁磁序二(Coll-II);,(d)奈尔反铁磁序(Nèel)Fig.6.Sketches of several magnetic orders in Fe-adsorbed biphenene monolayer:(a)Ferromagnetic order;(b)collinear anti-ferromagnetic order I;(c)collinear anti-ferromagnetic order II;(d)Nèel antiferromagnetic order.
3.7 用GGA+U 方法考虑电子关联修正后的计算结果
GGA+U方法能够用直接增加d 轨道在位库仑排斥能的方式修正d 电子的关联效应,比杂化泛函方法能得到更清晰的物理图像,又采用GGA +U方法计算了联苯烯吸附铁原子的吸附位点、电子结构及磁性,以进一步核实前面的计算结果.首先,通过线性响应方法自洽地得到了联苯烯吸附铁原子体系的U值[45].如图7 所示,蓝线和红线分别为自洽和非自洽情形下d 轨道电子数对微小HubbardU值的线性响应,斜率为k1和k2,则U=1/k1—1/k2=3.6 eV.另外,吸附体系中易于产生电极化和瞬态偶极相互作用,色散力对吸附构型起着重要的作用,因而在计算中采用DFT-D2 形式来引入范德瓦耳斯修正[46].在考虑电子关联和色散关联修正情况下,对图1(b)中联苯烯的各个吸附位点优化了铁原子吸附的几何结构,并用(1)式计算了吸附能.结果表明,空位吸附比桥位和定位吸附更稳定.空位吸附时,四环、六环、八环空位的吸附能分别为1.45,1.21,1.19 eV,故四环空位是最稳定的吸附位置,与前面计算的结论一致.图5(b)给出了GGA+U计算的四环空位的铁原子投影态密度,由图5(b)可以看出,自旋向上和向下的d 电子态发生更大的劈裂,自旋向上d 电子态处于更深的能量范围(—6.0 eV 至—4.0 eV).由图5(d)可以看出,自旋向下的电子态移到了费米能之上,这将导致更大磁矩出现,铁原子的磁矩为 4.87μB.另外,对于图6 中4 种磁序,也计算了相应的能量,列于表2 中.考虑关联效应后,奈尔反铁磁序的能量仍然最低,进一步确认了其铁磁奈尔序基态.

图7 自洽计算(蓝线)和非自洽计算(红线)铁原子d 电子数对Hubbard U 的响应.Fig.7.Response of d electron number of Fe atom adsorbed on biphenene network to Hubbard U in self-consistent and non-self-consistent calculations.
4 总结与展望
本文通过密度泛函理论框架下的第一性原理计算研究了联苯烯单层对铁原子的吸附情况,明确了联苯烯单层可能的吸附位点.通过吸附能和分子动力学的计算,证明了铁原子可以稳定的吸附于联苯烯单层四元环和六元环的空位上.铁原子与联苯烯单层存在明显的电荷转移,铁3d 电子态与碳2p 电子态有较强的轨道杂化,二者间能够形成稳定的化学键.铁原子在四元环空位吸附时,易磁化轴指向面外,磁矩大小约为1.81μB.相对于石墨烯而言,联苯烯是更优异的吸附载体,体现在两个方面,一是吸附能较大,二是吸附位点(四环空位)分散且间距合理,这导致吸附后能形成稳定的复合材料.联苯烯作为磁性材料,而且磁性原子排列规则,便于形成稳定的磁序;作为催化材料,催化位点稳固,分散合理.联苯烯的另一个优点是金属性,优良的导电性有利于作为分子导线、电极材料等电输运器件.金属性也有利于联苯烯吸附铁原子作为催化材料,有实验研究表明[53],金属态的电催化剂具有更为优异的电催化活性,其原因是电子在金属态的催化剂内部可以更快地传输.因此,本文结果确认了联苯烯单层是比石墨烯更好的铁原子吸附载体且体系有磁性,为研究新型吸附材料的电磁、输运、催化等特性提供了重要的基础信息.
猜你喜欢
物理学报(2022年17期)2022-09-14
中老年保健(2022年3期)2022-08-24
快乐学习报·教师周刊(2022年3期)2022-04-21
中小学班主任(2019年12期)2019-09-10
作文中学版(2018年1期)2018-11-28
农业工程技术·温室园艺(2017年10期)2017-12-02
新课程·中旬(2016年12期)2017-05-08
中学生数理化·高二版(2017年2期)2017-04-19
分析化学(2015年7期)2015-07-30
读者欣赏(2014年6期)2014-07-03
