
基于核磁共振和高分辨质谱技术确定艾叶中的白坚木皮醇△
2022-05-08 23:53陈阳江丹廖子蔚王玉孙代华陈志元
中国现代中药 2022年4期
陈阳,江丹,廖子蔚,王玉,孙代华,陈志元*
1.劲牌持正堂药业有限公司,湖北 黄石 435100;2.湖北省中药配方颗粒工程技术研究中心,湖北 黄石 435100
艾叶是菊科植物艾Artemisia argyiLevl.et Vant的干燥叶,也被称为“艾蒿”和“冰台”,是一种药食同源的植物资源,广泛生长于中国华南和华北等地[1]。艾叶具有多重功效,在传统功效上主要用于治疗痢疾、炎症、白血病等疾病。目前,对于艾叶的研究主要集中于黄酮类、多糖、生物碱等成分和乙醇提取物[2-3]。艾叶中存在大量的白坚木皮醇,但纯化方法研究较少,提纯较为困难且没有对艾叶中提取、纯化出的白坚木皮醇成分进行鉴别[4]。
白坚木皮醇(L-quebrachitol),又名2-O-甲基-L-(-)-肌醇(2-O-methyl-L-chiro-inositol)、白雀木醇,是一种天然存在、具有旋光活性的手性药物中间体,最早从南美洲的白坚木中发现。白坚木皮醇常作为一种优良的药物原料用于合成具有较高经济价值的光学活性肌醇衍生物[5]。白坚木皮醇药用疗效突出,现已被证明具有辅助治愈癌症、促进细胞增殖、辅助治疗糖尿病、抗菌和减轻胃损伤的作用,引起了制药领域学者的广泛关注[6-8]。迄今为止,白坚木皮醇的获取主要来自于天然橡胶乳清、荔枝和沙棘,其质量分数为0.2%~1.2%[9]。由于白坚木皮醇在上述原料中含量较低、提取液中同分异构体较多、生产过程需要使用大量有机溶剂等,导致高纯度白坚木皮醇无法工业化生产。笔者课题组利用结晶、重结晶的方法从艾叶药材中制备出高纯度白坚木皮醇,通过核磁共振(NMR)和高分辨-电喷雾离子源-质谱(HR-ESI-MS)数据确定结论的可靠性,并对白坚木皮醇的提取、纯化工艺进行了详细的描述,最后使用高效液相色谱-蒸发光散射检测器法(HPLC-ELSD)对多地区产艾叶进行白坚木皮醇含量分析,旨在为高纯度白坚木皮醇的工业化生产提供新方法,促进中药资源的高效利用。
1 材料
1.1 试药
艾叶药材田间直采于湖北、河南等地,经湖北师范大学张新潮副教授鉴定为菊科植物艾Artemisia.argyiLevl.et Vant.的干燥叶;95%乙醇(批号:A202010-1A)购于国药集团化学试剂有限公司;乙腈(色谱纯,批号:T202008-1G)购于赛默飞世尔科技有限公司;白坚木皮醇对照品(批号:G3002-130UN,纯度>99.54%)购于Sigma 公司;其余试剂均为分析纯。
1.2 仪器与耗材
1200 型高效液相色谱仪(安捷伦有限公司);AB135-S 型分析天平(梅特勒-托利多公司);Inertsil ODS-SP C18柱(岛津实验器材有限公司);AllChrom ELSD6000 型蒸发光散射器(赛默飞世尔科技有限公司);DSC-60PLUS 型差示扫描量热仪(日本岛津公司);WZZ-2S 型自动旋光仪(上海仪电物理光学仪器有限公司);DPX-500型核磁共振波谱仪(瑞士Bruker 公司);SYNAPT G2-S 型质谱仪(美国Waters 公司);0.22 μm 微孔滤膜(批号:G122011-2T)购于天津市津腾实验设备有限公司。
2 方法与结果
2.1 高纯度白坚木皮醇的制备
称取干燥的艾叶药材20 kg,加入15 倍质量的80%乙醇在常温下搅拌提取3 次,每次提取3 h,合并得到的提取液,然后70 ℃真空减压浓缩5 h 得到艾叶提取物浸膏800 g(样品A),在样品A 中加入2.5倍质量的纯净水并搅拌溶解,滤过得到滤液2.6 kg(样品B)和不溶性浸膏150 g(样品C),将样品B在65 ℃条件下真空减压浓缩3 h 得到艾叶水洗液浸膏320 g(样品D),加入5倍质量的90%乙醇溶解结晶得到褐色晶体74 g(样品E),将晶体分离后加入3倍质量的75%乙醇重结晶得到灰色晶体52 g(样品F),最后将晶体分离再加入3 倍质量的70%乙醇重结晶得到白色晶体36 g(样品G)。
2.2 溶液配制
2.2.1白坚木皮醇对照品溶液配制 精密称定白坚木皮醇对照品40 mg 于50 mL 的量瓶中,加纯水约40 mL,超声(250 W,53 kHz)溶解10 min 后放置至室温,用纯水溶解定容至刻度,摇匀。再精密吸取该溶液2.0、4.0、6.0、8.0、10.0 mL分别至10 mL量瓶中,加水至刻度,摇匀,分别过0.22 μm微孔滤膜,即得系列质量浓度的白坚木皮醇对照品溶液。
2.2.2供试品溶液配制 精密称定样品A、C、F、G 各约1.0 g 分别置于150 mL 锥形瓶中,加入30%乙醇100 mL,称定质量,超声(250 W,53 kHz)75 min 后,放置至室温,再称定质量,用30%乙醇补足减失的质量,摇匀,过0.22 μm 微孔滤膜,即得供试品溶液。
2.3 高纯度白坚木皮醇鉴定
2.3.1示差扫描量热法(DSC)测定熔点 分别精确称取样品G 和白坚木皮醇对照品1.0 mg,纯水溶解至终质量浓度为1.0 mg·mL-1,在氮气环境下注入到示差扫描量热仪中。
样品G 和白坚木皮醇对照品的DSC 曲线见图1。样品G 和白坚木皮醇对照品相对应的熔融峰温度分别为189.97 ℃和191.77 ℃。样品G 的熔点非常接近白坚木皮醇对照品,并且2 条曲线均具有非常尖锐的吸收峰且曲线重合度相似。说明样品G 纯度较高,极有可能是高纯度白坚木皮醇。

图1 样品G和白坚木皮醇对照品的DSC曲线
2.3.2比旋光度测定 分别精确称取样品G 和白坚木皮醇对照品1.0 mg,纯水溶解至终质量浓度为1.0 mg·mL-1,在20 ℃条件下注入到20 cm 的旋光管中测定旋光度。为了确保测量结果的准确性,所有实验均进行3 次,并通过公式(1)计算出比旋光度值。

式中α为旋光度值;[α]为比旋光度值;L为样品管的长度(dm);C为样品溶液的质量浓度(g·mL-1)。
结果显示,样品G 3 次旋光度平均值是0.147±0.002,白坚木皮醇对照品3 次旋光度平均值是0.148±0.001,经过计算分别得出样品白坚木皮醇对照品样品G和白坚木皮醇对照品的[α]基本一致,且这2种物质均为左旋。
2.3.3高分辨质谱条件 将样品G 溶解在甲醇中,终质量浓度为1.5 mg·mL-1,然后取样品1.5 μL 注入进样口。质谱条件为电喷雾离子源(ESI):正负离子模式;源喷雾电压为3.5 kV(正离子模式),3.0 kV(负离子模式);鞘气流量为15 L·min-1;辅助气流量为5 L·min-1;加热毛细管温度为350 ℃,毛细管电压为35 V。
高分辨质谱图见图2,由HR-ESI-MS 数据可以推断出m/z217.070 0[M+Na]+为准分子离子峰,m/z411.148 6[2M+Na]+为二聚体分子离子峰,样品G相对分子质量为194.079 0。
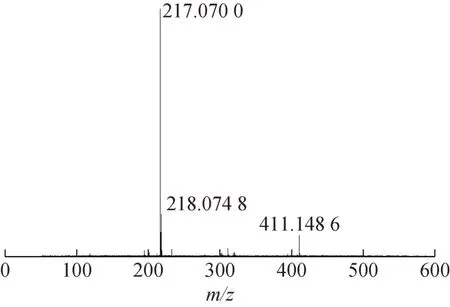
图2 样品G高分辨质谱图
2.3.4NMR 分析 样品G 的NMR 数据见图3。1HNMR(D2O,500 MHz)δ:3.34(1H,dd,J=3.0,9.6 Hz,H-2),3.39 (3H,s,H-OMe),3.55 (2H,m,H-3,4),3.68 (1H,dd,J=9.7,3.2 Hz,H-5),4.00 (1H,t,J=3.7 Hz,H-6),4.21 (1H,t,J=3.7 Hz,H-1);13C-NMR(D2O,125 MHz)δ:57.4 (Me),67.7 (C-1),80.7 (C-2),73.4(C-3),72.5(C-4),70.9(C-5),71.9(C-6)。
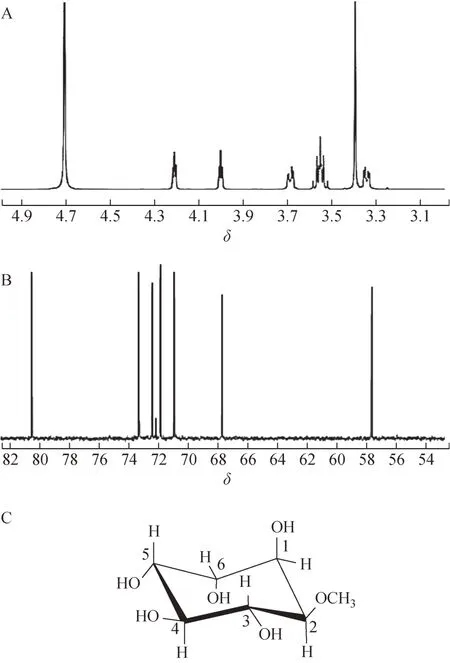
图3 样品G的NMR谱图及分子结构式
根据图3A 并结合NMR 数据可知,δH3.3~4.3存在6 个氢信号。氢信号之一分配给δH3.39 处的3个甲氧基氢,次甲基氢存在2 个未解析的信号,产生多重峰(δH3.55),而4 个氢信号归因于次甲基氢(δH3.34,3.68,4.00,4.21)。图3B 显示样品G的13C-NMR 谱图中扫描到7 个化学位移峰,包括δC67.7(C-1),80.7(C-2),73.4(C-3),72.5(C-4),70.9(C-5),71.9(C-6),57.4(Me)。将谱图中的数据与文献所报道的白坚木皮醇信息进行比较,结果表明样品G 的谱学数据与天然白坚木皮醇谱学数据相同[10]。综合分析以上实验结果,根据NMR 数据并结合质谱数据,推断样品G 的分子结构式见图3C,分子式为C7H14O6,样品G即为白坚木皮醇。
2.4 白坚木皮醇含量的测定
参照董铁山等[11]的方法,具体色谱条件如下:Inertsil ODS-SPC18色谱柱(150 mm×4.6 mm,5.0 μm);ELSD 漂移管温度为100 ℃;氮气流速为2 L·min-1;流动相为乙腈-水(9∶1);流速为0.2 mL·min-1;进样体积为1 μL;柱温为35 ℃。采用HPLC-ELSD 分别对2.1项下样品A、C、F和G进行白坚木皮醇含量测定。
4 个样品的HPLC-ELSD 图见图4,图4B 中有较多的杂峰,表明样品A含有较多的杂质,而图4C中没有出现代表白坚木皮醇的峰,表明白坚木皮醇转移到了样品B中,不溶性物质样品C基本不含有白坚木皮醇。图4D中出现的杂峰极少,表明样品E经过1次重结晶得到的样品F中白坚木皮醇的杂质减少,而在图4E中基本没有杂峰出现,表明样品E经过2次重结晶后纯度接近对照品。通过面积归一化法分别计算样品A、C、F 和G 中白坚木皮醇的含量分别为29.53%、0.20%、83.24%、99.18%,上述数据充分证明了本研究中制备高纯度白坚木皮醇的工艺是合理的。

图4 4个样品的HPLC-ELSD图
2.5 不同产地艾叶中白坚木皮醇含量的测定
艾叶原料前处理方式与2.2.2 项下供试品溶液配制方式相同。按照2.4 项下HPLC-ELSD 分别测定了来自河南和湖北9 个产地艾叶中白坚木皮醇的含量。测定结果见表1。
表1展示了9个不同产地艾叶中白坚木皮醇的质量分数。从表1 中可以看出所有的艾叶样品中都含有白坚木皮醇,其质量分数为16.7~34.9 mg·g-1。2020年6月8日从湖北省麻城市采收的艾叶中白坚木皮醇的含量最高,而不同月份在麻城市采收的艾叶中白坚木皮醇的含量有一定波动性,这说明艾叶的产地及采收时间可能对艾叶中白坚木皮醇的含量有影响,这个研究结果与Yang 等[12]的研究结果相似。因此,笔者课题组将在下一步工作中研究艾叶采收地区和采收时间对白坚木皮醇含量的影响,以期找到艾叶的最佳产地和采收时间。
表1 不同产地艾叶中白坚木皮醇含量检测结果(,n=3)
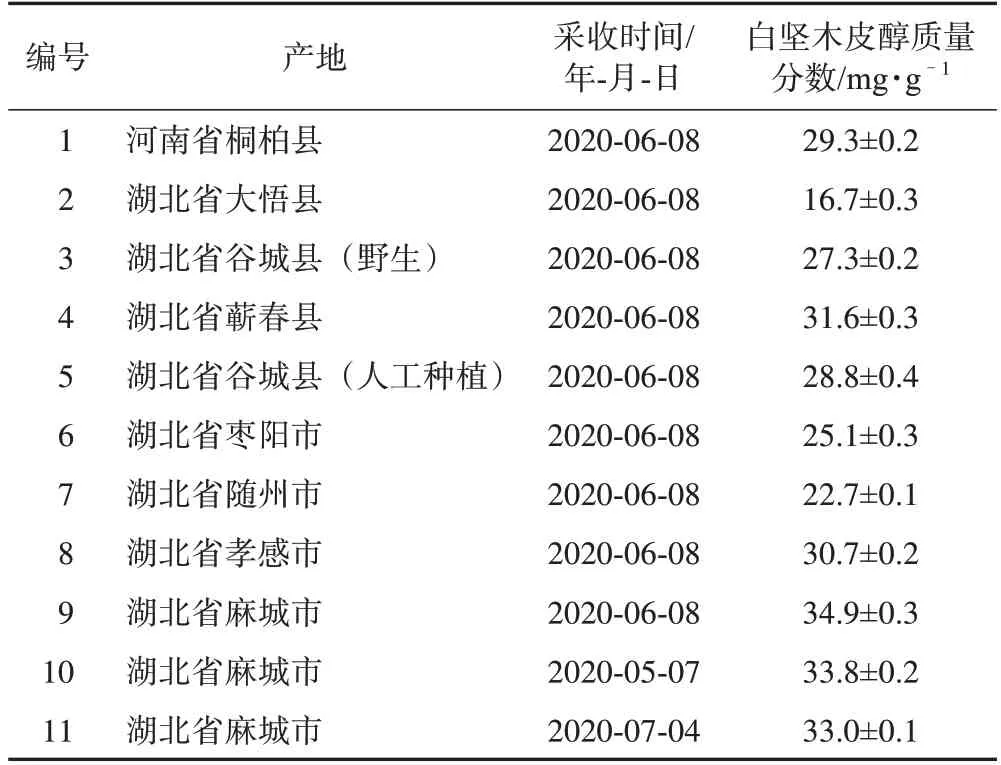
表1 不同产地艾叶中白坚木皮醇含量检测结果(,n=3)
2.6 数据处理
DSC 测定、比旋光度测定及不同产地艾叶中白坚木皮醇含量的测定均进行3 次平行实验,并采用SPSS 20.0进行数据处理,数据以()表示。
3 讨论与结论
白坚木皮醇先后在白坚木、巴西橡胶、荔枝、龙眼及沙棘果中被发现,因其可以作为无毒、无害原料,合成用于抗癌、辅助治疗阿尔茨海默病、治疗糖尿病的手性药物而受广大医者的青睐。董铁山等[13]利用高效液相色谱-二极管阵列检测器法(HPLC-DAD)证明了天然橡胶乳清中存在白坚木皮醇,并利用乙酸和氢氧化钙脱除天然乳清中的杂蛋白,最后使用大孔树脂得到粗白坚木皮醇晶体,工艺过程操作复杂、产率低且需使用较多的有机溶剂。综合文献还未发现一种简单、高效、无毒的制备高纯度白坚木皮醇的方法,本研究将为制备高纯度白坚木皮醇提供一个新思路,从而提高艾叶药材资源的附加值。
本研究利用醇提、水洗、析晶和重结晶的方法制备了高纯度白坚木皮醇,采用DSC、比旋光度法、NMR 和HR-ESI-MS 鉴定艾叶中分离、纯化出的白色晶体为高纯度白坚木皮醇,并在此基础上对样品G 和来自不同地区的艾叶药材进行白坚木皮醇含量测定。结果显示,样品G 与白坚木皮醇对照品的熔点和比旋光度相似,NMR 和HR-ESI-MS 数据与已报道的白坚木皮醇相同,从而鉴定得到的白色晶体样品G 为高纯度白坚木皮醇。HPLC-ELSD 法测定出样品G 的纯度高达99.18%,且艾叶药材中白坚木皮醇的含量与产地、采收时间有关。下一步,笔者课题组将就艾叶采收地区和采收时间对白坚木皮醇含量的影响进行深入的研究,对白坚木皮醇的药理作用进行发掘,从而扩宽白坚木皮醇的应用范围。
猜你喜欢
中老年保健(2022年6期)2022-08-19
纺织标准与质量(2022年1期)2022-07-12
当代水产(2022年4期)2022-06-05
福建轻纺(2022年4期)2022-06-01
中国药学药品知识仓库(2022年8期)2022-05-09
军民两用技术与产品(2021年10期)2021-11-25
文萃报·周二版(2021年9期)2021-03-10
中国食品(2020年9期)2020-05-26
食品安全导刊(2017年12期)2018-01-04
食品界(2017年7期)2017-08-24
