
木论喀斯特常绿落叶阔叶混交林土壤真菌群落多样性特征
2022-05-14 03:13谷俊锟曾馥平宋同清彭晚霞苏樑杜虎
生态科学 2022年3期
谷俊锟, 曾馥平 宋同清 彭晚霞 苏樑 杜虎*
木论喀斯特常绿落叶阔叶混交林土壤真菌群落多样性特征
谷俊锟1,2,3, 曾馥平1,2, 宋同清1,2, 彭晚霞1,2, 苏樑1,2, 杜虎1,2,*
1. 中国科学院亚热带农业生态研究所亚热带农业生态过程重点实验室, 湖南长沙 410125 2. 中国科学院环江喀斯特生态系统观测研究站/广西喀斯特生态过程与服务重点实验室, 广西环江 547100 3. 湖南农业大学资源环境学院, 湖南长沙 410125
土壤微生物在森林生态系统中起着重要作用, 为了解喀斯特常绿落叶阔叶混交林土壤真菌多样性, 采用Illumina Hiseq高通量测序技术对木论喀斯特森林土壤真菌群落多样性进行了初步研究。研究结果表明: 在82个采样点内, 随着采样点增加, 检测出不同分类水平的土壤真菌类群逐步增多; 土壤样点数达到82个时, 检测出的土壤真菌类群达6门32纲126目336科886属和132616个种(OTU); 82个土壤样品中所检测出的真菌类群平均有5.41门22.13纲71.09目144.89科216.66属和1256.44个OTU, 其中门、纲、目分类水平上的优势类群(所占比例)分别为子囊菌门()(50.09%)、接合菌纲()(34.54%)、被孢霉目()(22.65%)。取样数量对所检测土壤真菌类群数据有显著影响, 在门、纲、目、科的分类水平上, 1个、15个、48个、73个样品基本上可以代表82个样品检测出来不同分类水平的真菌多样性; 在属和OTU的分类水平上, 当样品数达到82个时检测出来的响应类群数仍在不断增加。该研究为进一步了解喀斯特森林土壤真菌多样性与植物多样性和生境的关系提供了基础。
土壤真菌多样性; 高通量测序; 采样数; 森林; 喀斯特生态系统
0 前言
微生物是土壤最活跃的组成, 在土壤中储存了数量庞大微生物资源[1, 2]。土壤微生物参与了土壤发生、发展、发育的全过程, 在物质循环、能量转换以及污染物降解等过程中都发挥着重要作用[3]。真菌作为土壤微生物中的重要组成部分, 可有效降解土壤中复杂组分和凋落物, 驱动养分循环和能量流动, 还可反映土壤病理过程, 是陆地生态系统的重要健康指标[4, 5], 并且和植物、细菌之间维持着重要的共生关系[6]。大量研究发现, 森林生态系统具有极其丰富的微生物资源, 且由于微生物结构相对简单, 能对环境变化做出快速而灵敏的反应。相对于土壤细菌, 虽然森林土壤真菌的研究受到的关注偏少, 但真菌在森林生态系统中对于促进宿主植物对矿物质吸收、稳固和改善土壤结构的作用不容忽视[7-9]。在亚热带不同植被的影响下, 随着生态环境改善、植被的恢复, 土壤真菌群落多样性逐渐增加[10]。中温带气候下不同林型的土壤真菌群落结构存在较大差异。在寒温带气候下, 森林植被类型、土壤环境因子和林下植被对土壤真菌群落结构影响显著[11, 12]。不同植被类型凋落物通过改变土壤理化性质进而影响土壤真菌多样性[13]。充分了解森林生态系统土壤真菌群落多样性对认识生态系统结构、功能与过程, 评价生态系统服务等具有重要的意义。在热带和温带, 经大量研究发现, 土壤真菌门类群含量一致丰富的是子囊菌门[14-16], 土壤真菌属类群丰富的是被孢霉属[17]。据估算, 全球范围内真菌种数可达80-510万, 而目前已知大约有10万种[5]。因此更加全面的反映微生物信息显得尤为重要。随着分子生物学技术的发展, 从早先的平板培养法发展到高通量测序技术的应用, 使得更为简单、快速、准确的获取土壤微生物信息成为可能[2]。获得一定数量且具有代表性的森林土壤样品是森林土壤微生物群落调查的首要工作。在目前的研究当中对每个样地或样方内需要采集多少数量的样品并没有统一标准[18]。在考虑代表性和可操作性前提下, 究竟多少个土壤样品才能充分反映森林土壤微生物群落信息?这是一个基础但重要问题。
位于我国西南喀斯特地区的广西木论国家级自然保护区与毗邻的贵州茂兰国家级自然保护区连片保存着目前世界上连片面积最大、保存最完好、原生性最强的喀斯特非地带性植被——喀斯特常绿落叶阔叶混交林。该区域也是我国生物多样性3个特有分布中心之一, 且位于我国植物区系相汇交错区和交接过渡带, 其植被和生境呈现高度的异质性[19, 20], 是进行喀斯特地质背景下生物多样性相关研究的理想场所。本研究基于Illumina HiSeq测序平台对位于木论国家级自然保护区内25 ha大样地土壤真菌多样性进行了初步研究, 分析了土壤真菌门、纲、目、科、属等不同分类水平优势类群和各类群相对丰度, 并探讨了取样数量对土壤真菌多样性检测结果的影响, 以及在该生境内多少个土壤样品可以代表不同分类水平的真菌多样性, 其结果将有助于进一步回答土壤真菌多样性及其与植物多样性的关系。
1 材料与方法
1.1 研究区概况
木论国家级自然保护区位于广西环江毛南族自治县西北部, 地理坐标为107°54´01—108°05´51 E, 25°07´01—25°12´22 N之间, 属于中亚热带石灰岩区常绿落叶阔叶混交林生态系统, 是我国生物区系相汇交错区和交接过渡的中心, 生境异质性极高, 区系成分复杂, 生物种类丰富, 生态环境脆弱, 是目前世界上喀斯特地貌区幸存连片面积最大、保存最完好、原生性最强的喀斯特森林。该区域为中亚热带季风气候, 年平均气温19.3℃, 年均降水量1529 mm。区内土壤主要为非地带性石灰土。2014年参照CTFS(Center for Tropical Forest Sciences)森林样地建设技术要求在该保护区内建成了25 ha (500 m×500 m)森林动态监测样地。
1.2 土壤样品采集
监测样地按网格法划分成了625个20 m×20 m样方, 2016年秋季在监测样地中相对均匀地选取了82个样方进行土壤样品采集(图1), 在各样方中心点附近用土钻采集5个0—10 cm表层土壤混合成一个样品, 样品去除根系、石头等杂质后, -80 ℃保存用于DNA提取和后续实验分析。
1.3 DNA提取、扩增和高通量测序
土壤微生物宏基因组DNA提取采用的专用试剂盒(FastDNA®SPIN Kit for Soil, MP)。采用引物ITS5-1737F(GGAAGTAAAAGTCGTAACAAGG), ITS2-2043R(GCTGCGTTCTTCAT CG A TGC)对真菌ITS基因的ITS1区进行PCR扩增。PCR反应体系30 μL: 15 μL Phusion Master Mix(2×), 3 μL(6μM) Primer(2μM), 10 μL(5—10 ng)gDNA(1 ng/μL), 2 μL H2O[21]。PCR反应条件为: 98℃预变性1 min; 30个循环包括(98 ℃, 10 s; 50 ℃, 30 s; 72 ℃, 30 s); 72 ℃, 5 min。依据PCR产物浓度, 将所有扩增成功的PCR产物等量混合, 再经定量等质量控制后, 将质量合格的PCR产物进行DNA文库的构建, 采用Illumina Hiseq平台进行双末端250 bp测序(委托诺禾致源生物信息科技有限公司完成)。
1.4 高通量测序数据处理与统计分析
根据Barcode序列和PCR扩增引物序列从下机数据中拆分出各样品数据, 截去Barcode和引物序列后使用FLASH(V1.2.7, http://ccb.jhu.edu/software/ FLASH/)[22]对每个样品的reads进行拼接, 过滤掉低质量的序列。使用Qiime(V1.7.0, http://qiime.org/ scripts/split_libraries_fastq.html)[23]的Tags质量控制流程做聚类及多样性分析。其中, 利用Uparse软件(Uparse v7.0.1001, http://drive5.com/uparse/)[24]对所有样品进行聚类, 依据惯例以97%的一致性将序列聚类成为OTUs[25], 依据其算法原则, 筛选出OTUs中出现频数最高的序列作为OTUs的代表序列, 用RDP对其进行系统分类[26]。再用QIIME进行单样品组成分析[23], 得到样品在不同分类水平上的真类群组成及相对丰度的数据, 将各分类水平: Phylum(门)、Class(纲)、Order(目)、Family(科)、Genus(属)上最大丰度排名前10的物种, 生成物种相对丰度图[27]。

图1 木论森林动态监测样地土壤真菌采样点位置
Figure 1 Location of soil fungi sampling points in Mulun Forest Dynamic Monitoring Plot
在上述基础上制作采样点数–土壤真菌类群数曲线。具体的做法是: 从82个样品中随机抽取1个样品, 记下这个样品中包含的真菌类群数目; 然后从剩下的81个样品中再随机抽取1个样品数据, 和第一个抽到的样品数据合并, 记下这2个样品中包含的真菌类群数目; 依此类推, 直至采样点的数目达到最大; 重复99次取平均值, 制作采样点数–土壤真菌类群数曲线。
本研究其他数据分析与图形制作使用Origin 2016和R3.5.1软件(R-development Core Team, 2018)完成。
2 结果与分析
2.1 土壤真菌分类
对木论样地的82个土壤样品进行高通量测序, 共产生132616个OTU。与数据库比对不上的OTU即未分配的部分(Unassigned)约占总数的4.414%。82个土壤样品中平均有1256.44 ±493.32个OUT, 其中单个样品最多为3359个、最少为653个, 分属于6门32纲126目336科886属, 真菌分类群详细信息见附录1(不包含未分配的部分; 各个分类水平上未有明确分类名称信息的也作为一个计入在内)。
82个土壤样品中平均有真菌5.41 ±0.51门, 其中单个样品最多的有6门, 最少的有5门(图2)。相对丰度最高的5个门所占比例总和达到了99.56%, 分别是子囊菌门(Ascomycota)(50.09%)、接合菌门(Zygomycota)(42.13%)、担子菌门(Basidiomycota) (6.86%)、壶菌门(Chytridiomycota)(6.01%)和球囊菌门(Glomeromycota)(0.26%)(图3)。
82个土壤样品中平均有真菌22.13 ±2.43纲, 其中单个样品最多的有26纲, 最少的有18纲(图2)。相对丰度最高的5个纲所占比例总和为71.97%, 分别是接合菌纲(Zygomycota)(34.54%)、座囊菌纲(Dothideomycetes) (15.51%)、粪壳菌纲(Sordariomycetes)(14.04%)、伞菌纲(Agaricomycetes)(4.84%)和散囊菌纲(Eurotiomycetes)(3.05%)(图3)。
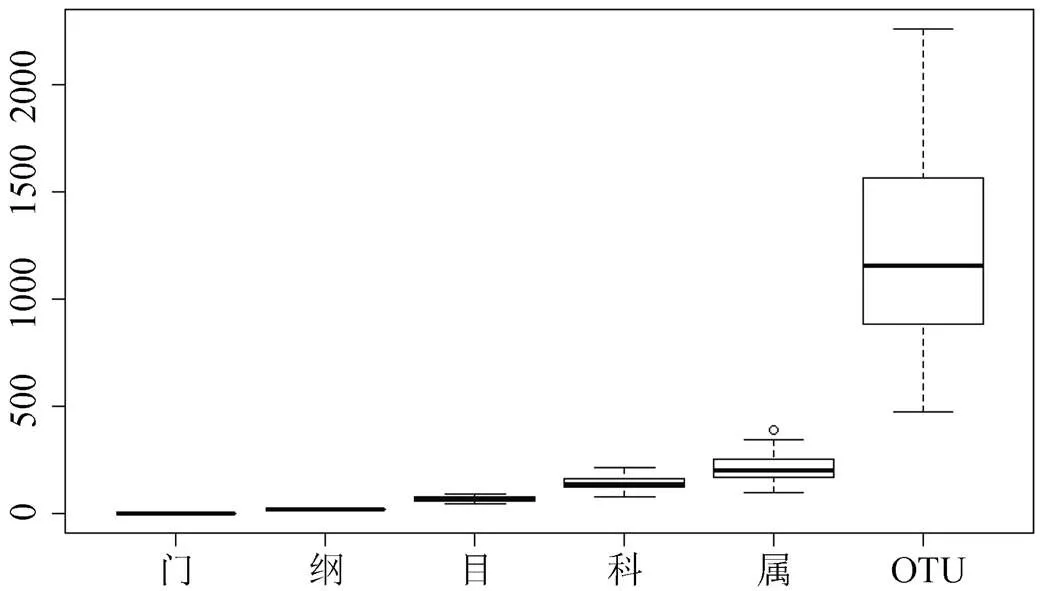
图2 不同分类水平上真菌数量
Figure 2 The quantity of soil fungal at different taxonomical levels
82个土壤样品中平均有真菌70.09±10.82目, 其中单个样品最多的有94目, 最少的有48目(图2)。相对丰度最高的5个目所占比例总和为39.42%, 分别是被孢霉目(Mortierellales)(22.65%)、肉座菌目(Hypocreales)(7.31%)、格孢菌目(Pleosporales) (4.21%)、炭角菌目(Xylariales)(2.71%)和伞菌目(Agaricales)(2.56%)(图3)。
82个土壤样品中平均有真菌144.89±30.02科, 其中单个样品最多的有219科, 最少的有83科(图2)。相对丰度最高的5个科所占比例总和为14.59%, 分别是被孢霉科(Mortierellaceae)(5.07%)、格孢菌科(Incertae_sedis_Pleosporales)(3.16%)、发菌科(Trichocomaceae)(2.22%)、炭角菌科(Incertae_sedis_ Xylariales)(2.09%)和古生菌科(Archaeorhizomycetaceae)(2.05%)(图3)。
82个土壤样品中平均有真菌216.66±65.73属, 其中单个样品最多的有392属, 最少的有99属(图2)。相对丰度最高的5个属所占比例总和为9.28%, 分别是被孢霉属(Mortierella)(3.68%)、小画线壳属(Monographella)(1.93%)、白粉菌属Microidium (1.23%)、周刺座霉属(Volutella)(1.23%)和青霉属(Penicillium)(1.21%)(图3)。
2.2 取样数对土壤真菌多样性的影响
在各个分类水平上, 随着取样点的增加, 检测出的土壤真菌类群的数目也在不断增加。当取样点达到82个时, 检测出的土壤真菌类群的数目为最大, 分别为6门32纲126目336科886属132616个OUT(图4)。
在门的分类水平上, 当取样点数为1个时, 约能检测到5个门; 取样点数在2—82区间时, 门数仅增加1个达到6个。
在纲的分类水平上, 取样点数在1—8区间时, 检测到的真菌纲数量随着取样点数量增加而快速增加; 取样点数超过8后, 纲的数量增速减慢; 当取样点数为15个时, 约能检测到31个纲; 取样点数在16—82区间时, 纲数仅增加1个达到32个。
在目的分类水平上, 取样点数在1—25区间时, 检测到的真菌目数量随着取样点数量增加而快速增加; 取样点数超过25后, 目的数量增速减慢; 当取样点数为48个时, 约能检测到125个纲; 取样点数在49—82区间时, 目数仅增加1个达到126。
在科的分类水平上, 取样点数在1—52区间时, 检测到的真菌科数量随着取样点数量增加而快速增加;
取样点数超过52后, 科的数量增速减慢; 当取样点数为73个时, 约能检测到335个目; 取样点数在74—82区间时, 科数仅增加1个达到336。
在属的分类水平上, 取样点数在1—70区间时, 检测到的真菌属数量随着取样点数量增加而快速增加; 取样点数超过70后, 属的数量增速减慢, 但增速仍较大, 且属的增速大于门、纲、目、科的增速。
在OTU的分类水平上, 当取样点数在1—82区间时, 检测到的真菌OTU数量随着取样点数量增加一直呈现出较快增加的趋势(图4)。
3 讨论
采用Illumina公司Hiseq测序仪对地处中亚热带季风气候区的木论喀斯特常绿落叶阔叶混交林土壤真菌多样性进行初步研究, 共产生132616个OUT, 分属于6门32纲126目336科886属。研究结果列出了木论样地土壤真菌各个不同分类水平上的优势类群及其相对丰度。其中,是最丰富的真菌类群, O'Brien 等[28]研究表明森林土壤 ITS 文库中的和序列比例(46%和41%)相差不大, 而和占ITS克隆文库序列的1.5%和0.5%。但本研究中序列仅占6.86%, 而序列占了 42.13%。这与 O'Brien的研究结果有一定的差异, 而Lienhard 等[29]在老挝热带草地中对真菌群落进行研究发现, 草地中真菌群落在门水平上的比例较高, 这与本研究结果一致。这可能由于和都属于腐生营养土壤真菌, 在土壤真菌群落中占主导地位。但在其他有关森林土壤中真菌群落的研究中发现,在真菌群落中的比例较高, 这可能是由于土壤质地和植被类型产生的[30]。
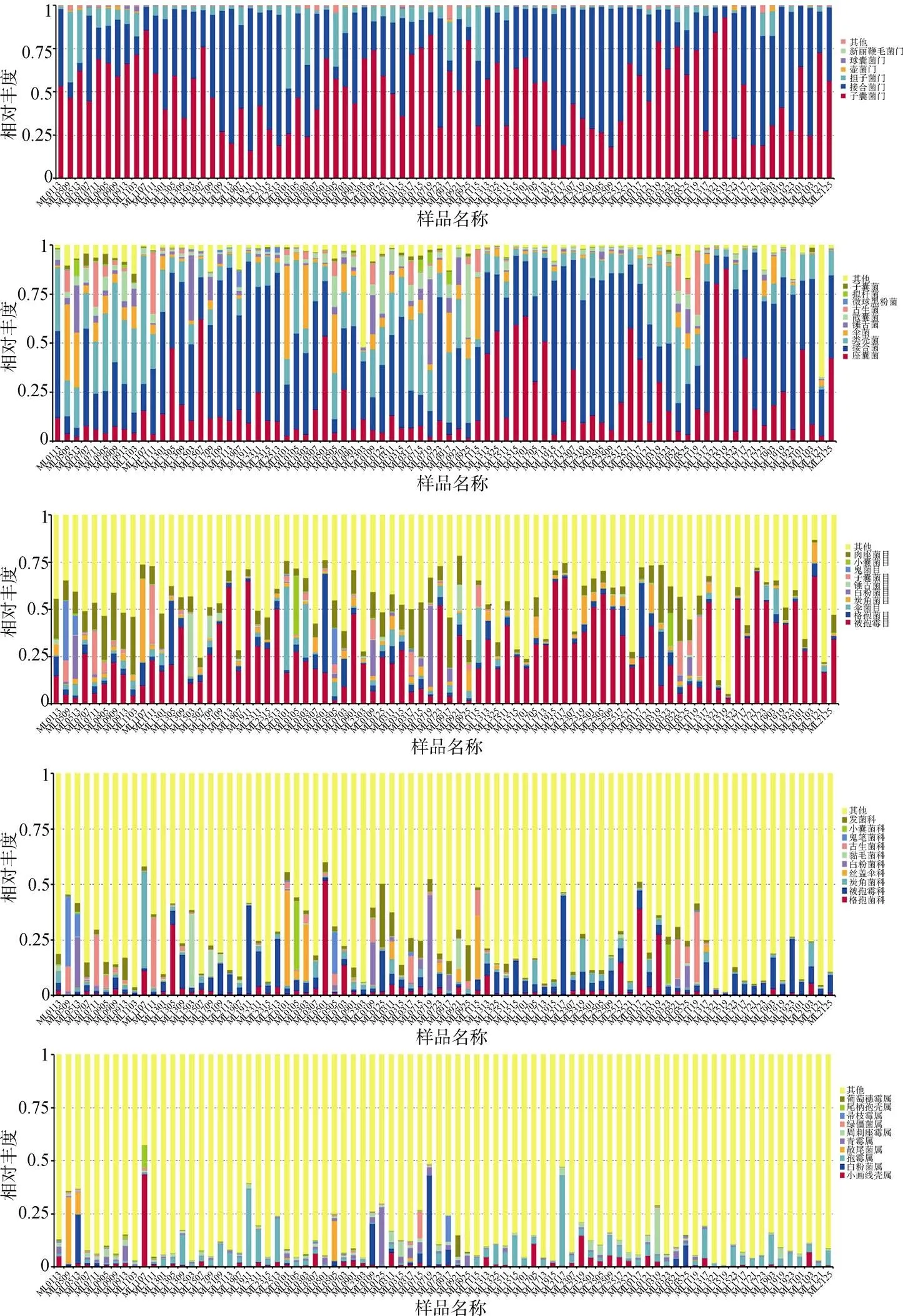
图3 木论大样地土壤样品在不同水平上真菌优势类群及其相对丰度
Figure 3 Dominant groups of fungi and their relative abundance at different levels in soil samples from the Mulun large plot.

图4 不同分类水平上真菌类群数随采样点数增加的稀疏曲线
Figure 4 Rarefaction curves of the observed fungi quantity at different taxonomic levels varying with number of samples in the study area
在某种植被类型、生境类型或是某个生态系统, 所需多少土壤样品才能较好地反映其微生物多样性水平, 依然是一个值得探究的问题。但在采样数量一定时, 研究人员大多还是将数个取样点土样混合后成一个样品可增加样品的代表性[31,32]。本研究中, 在不同的分类水平上, 随着取样点的增加, 检测出的土壤真菌类群的数目呈现出不断增加的趋势, 且增速不同, 但逐渐变缓。当取样点达到82个时, 检测出的土壤真菌类群的数目达到最大, 这说明当取样点数较少时, 可能存在较多的真菌类群没有被检测出来的情况。特别是在OTU的水平上, 当样品数量达到82个时, 真菌OTU数量仍呈现出较快增长的趋势, 由此推测, 如果样品数量增加较多, 被检测出来的真菌OTU数量还会有较大增加, 今后的试验进一步在木论喀斯特常绿落叶阔叶混交林中, 探究取样数目与土壤真菌类群数目在属和OTU水平上的联系。在以往对草地、农田或是在森林等土壤微生物研究中, 均应重视取样数量对于研究结果的影响[33-35]。取样数量除了取决于研究目的和研究深度外, 实验研究成本也会对其产生一定影响, 因为高通量测序单个样品所需经费相对较高, 因此, 我们在实际研究当中应对取样数量进行权衡, 既要能满足研究目的、反应研究对象的微生物多样性水平, 又要能兼顾到研究费用。除此之外, 检测出的真菌类群也会受到文库的构建和测序深度的选择等因素的影响。因此, 库容和测序深度的增加, 也有可能检测出来更多的真菌类群。还有, 测序区间的选择也是影响研究结果的一个因素, 例如: 测序的高变区不同, 其分类信息的精确性也会不一样。
4 结论
木论喀斯特常绿落叶阔叶混交林土壤真菌分属于6门32纲126目336科886属。相对丰度最高的2个门分别是子囊菌门、接合菌门, 其所占比例总和达到了92.22%; 纲类水平下, 以接合菌纲最高, 其次是座囊菌纲和粪壳菌纲, 三者所占比例总和达到了64.09%; 目类水平下, 相对丰度最高的是被孢霉目, 其占比为22.65%, 其余各目所占比例均低于10%; 相对丰度最高的5个科分别是被孢霉科、格孢菌科、发菌科、炭角菌科和古生菌科, 其所占比例为14.59%; 相对丰度最高的5个属分别是被孢霉属、小画线壳属、白粉菌属、周刺座霉属和青霉属, 其所占比例总和为9.28%。
随着采样点数量的增加, 不同分类土壤真菌水平有不同程度的增加, 在门、纲、目、科的分类水平上, 1个、15个、48个、73个样品基本上可代表全部样品检测出来不同分类水平的细菌多样性信息; 而属和OUT数仍随样品数量增加而增加, 未呈现平缓趋势, 说明在该生境内, 继续增加取样数目, 属和OTU数目还会有明细增加, 今后可通过补充采样来进一步了解该森林土壤真菌属和OUT特征。
致谢:感谢陈莉、胡芳同学在野外土样采集中的帮助, 感谢木论国家级自然保护区工作人员在野外工作中的支持和帮助。
[1] DEQUIEDT S, SABY N P A, LELIEVRE M, et al. Biogeographical patterns of soil molecular microbial biomass as influenced by soil characteristics and management[J]. Global Ecology and Biogeography, 2011, 20(4): 641–652.
[2] 宋长青, 吴金水, 陆雅海, 等. 中国土壤微生物学研究10年回顾[J]. 地球科学进展, 2013, 28(10): 1087–1105.
[3] 贺纪正, 王军涛. 土壤微生物群落构建理论与时空演变特征[J]. 生态学报, 2015, 35(20): 6575–6583.
[4] 盛玉钰, 丛静, 卢慧, 等. 神农架国家公园林线过渡带土壤真菌多样性[J]. 生态学报, 2018, 38(15): 5322–5330.
[5] TEDERSOO L, BAHRAM M, POLME S, et al. Global diversity and geography of soil fungi[J]. Science, 2014, 346,10.1126/science.1256688 .
[6] 陈晓, 刘勇, 李国雷, 等. 土壤真菌研究方法及人为干扰对森林土壤真菌群落影响研究进展[J]. 世界林业研究, 2011, 24(5): 7–12.
[7] 王楠楠, 杨雪, 李世兰, 等. 降水变化驱动下红松阔叶林土壤真菌多样性的分布格局[J]. 应用生态学报, 2013, 24(7): 1985–1990.
[8] VORISKOVA J, BRABCOVA V, CAJTHAML T, et al. Seasonal dynamics of fungal communities in a temperate oak forest soil[J]. The New phytologist, 2014, 201(1): 269–278.
[9] XIAO Wenya, ZHAO Jiahao, YAN Xinlin, et al. Tree Diversity Determines the Diversity of the Taxonomic and Functional Structure of the Fungal Community in Forest Litter in Southern China[J]. Forest Science, 2019, 65(1): 40–47.
[10] 张腾升. 亚热带不同植被恢复阶段植物与土壤微生物多样性特征及其相关关系[D]. 南昌: 南昌工程学院, 2019.
[11] 隋心, 杨立宾, 崔福星, 等. 汤旺河国家公园红松林土壤真菌多样性研究[J]. 中国农学通报, 2019, 35(22): 84–90.
[12] 乔沙沙, 周永娜, 柴宝峰, 等. 关帝山森林土壤真菌群落结构与遗传多样性特征[J]. 环境科学, 2017, 38(6): 2502– 2512.
[13] 吴佳伟, 杨瑞, 王勇, 等. 贵州草海流域三种不同植被类型根际土壤真菌结构组成和多样性[J]. 菌物学报, 2020, 39(7): 1250–1262.
[14] 周玉杰, 李建华, 张广宇, 等. 基于高通量测序的橡胶林土壤真菌多样性及群落组成分析[J]. 南方农业学报, 2018, 49(9): 1729–1735.
[15] 叶文雨, 谢序泽, 许钰滢, 等. 基于高通量测序技术分析2种菌草根际土壤真菌群落多样性[J]. 热带作物学报, 2020, 41(3): 556–563.
[16] 邓娇娇, 朱文旭, 张岩, 等. 辽西北风沙区不同人工林土壤真菌群落结构及功能特征[J]. 林业科学研究, 2020, 33(1): 44–54.
[17] 武俊男, 刘昱辛, 周雪, 等. 基于Illumina MiSeq测序平台分析长期不同施肥处理对黑土真菌群落的影响[J]. 微生物学报, 2018, 58(9): 1658–1671.
[18] 陈莉, 宋同清, 王华, 等. 木论喀斯特常绿落叶阔叶混交林土壤细菌多样性及其最优采样数[J]. 生态学报, 2019, 39(09): 3287–3296.
[19] DU Hu, HU Fang, ZENG Fuping, et al. Spatial distribution of tree species in evergreen-deciduous broadleaf karst forests in southwest China[J]. Scientific reports, 2017, 7(1): 15664.
[20] 兰斯安, 宋敏, 曾馥平, 等. 木论喀斯特森林木本植物多样性垂直格局[J]. 生态学报, 2016, 36(22): 7374–7383.
[21] 毛海萍, 袁开, 金仁耀, 等. 基于传统分离培养和高通量测序分析市售咸鳓鱼中微生物多样性[J]. 食品研究与开发, 2019, 40(21): 193–201.
[22] MAGOC T, SALZBERG S L. FLASH: fast length adjustment of short reads to improve genome assemblies[J]. Bioinformatics (Oxford, England), 2011, 27(21): 2957–2963.
[23] CAPORASO J G, KUCZYNSKI J, STOMBAUGH J, et al. QIIME allows analysis of high-throughput community sequencing data[J]. Nature Methods: Techniques for life scientists and chemists, 2010, 7(5): 335–336.
[24] EDGAR R C. UPARSE: highly accurate OTU sequences from microbial amplicon reads[J]. Nature methods, 2013, 10(10): 996–998.
[25] STACKEBRANDT E, GOEBEL B M. Taxonomic Note: A Place for DNA-DNA Reassociation and 16S rRNA Sequence Analysis in the Present Species Definition in Bacteriology[J]. International Journal of Systematic Bacteriology, 1994, 44(4): 846–849.
[26] MAIDAK B L, COLE J R, LILBURN T G, et al. The RDP-II (Ribosomal Database Project)[J]. Nucleic acids research, 2001, 29(1): 173–174.
[27] 赵爱花, 杜晓军, 臧婧, 等. 宝天曼落叶阔叶林土壤细菌多样性[J]. 生物多样性, 2015, 23(5): 649–657.
[28] O'BRIEN H E, PARRENT J L, JACKSON J A, et al. Fungal community analysis by large-scale sequencing of environmental samples[J]. Applied and environmental microbiology, 2005, 71(9): 5544–5550.
[29] LIENHARD P, TERRAT S, PREVOST-BOURE N C, et al. Pyrosequencing evidences the impact of cropping on soil bacterial and fungal diversity in Laos tropical grassland[J]. Agronomy for Sustainable Development, 2014, 34(2): 525–533.
[30] LIM Y W, KIM B K, KIM C M, et al. Assessment of soil fungal communities using pyrosequencing[J]. Journal of microbiology (Seoul, Korea), 2010, 48(3): 284–289.
[31] FIERER N, JACKSON R B. The diversity and biogeography of soil bacterial communities[J]. Proceedings of the National Academy of ences of the United States of America, 2006, 103(3): 626–631.
[32] LI Hui, YE Dandan, WANG Xugao, et al. Soil bacterial communities of different natural forest types in Northeast China[J]. Plant and Soil, 2014, 383(1/2): 203–216.
[33] GRIFFITHS R I, THOMSON B C, JAMES P, et al. The bacterial biogeography of British soils[J]. Environmental Microbiology, 2011, 13(6): 1642–1654.
[34] JONES R T, ROBESON MS, LAUBER C L, et al. A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses[J]. The ISME journal, 2009, 3(4): 442– 453.
[35] LAUBER L C, HAMADY M, KNIGHT R, et al. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale[J]. Applied and environmental microbiology, 2009, 75(15): 5111–5120.
Soil fungal community composition in the Mulun karst evergreen and deciduous broad-leaved mixed forest
GU Junkun1,2, ZENG Fuping1,2, SONG Tongqing1,2, PENG Wanxia1,2, SU Liang1,2, DU Hu1,2,*
1. Key Laboratory of Agro-ecological Processes in Subtropical Region, Institute of Subtropical Agriculture, Chinese Academic of Sciences, Changsha, Hunan 410125, China 2. Guangxi Key Laboratory of Karst Ecological Processes and Services, Huanjiang Observation and Research Station of Karst Ecosystem, Huanjiang, Guangxi 547100, China 3. College of Resources and Environment, Hunan Agricultural University, Changsha 410125, China
Soil microbes play essential roles in forest ecosystems. To understand the diversity of soil fungal in karst forest, we investigated soil fungal community structure in the Mulun karst evergreen and deciduous broad-leaved mixed forest using the technology of Illumina Miseq. The results showed that in the 82 soil sampling sites, the number of fungal taxa detected at different classification levels increased with the increasing number of sampling points. When all 82 samples were considered, the number of relative fungal groups include 6 phyla, 32 classes, 126 orders, 336 families, 886 genera, and 132616 operational taxonomic units (OTUs). The mean values of relative fungal taxa in the 82 samples were 5.41 phyla, 22.13 classes, 71.09 orders, 144.89 families, 216.66 genera, and 1256.44 OTUs. At the classification level of phylum, class, and order, the dominant groups were(50.09%),(34.54%),(22.65%), respectively. These preliminary results show that the soil has a relatively high level of fungal diversity in the karst evergreen and deciduous broad-leaved mixed forest, which lays the foundation for further studies on the relationship between soil fungal diversity and plant diversity and other related scientific questions.
soil fungal diversity; high-throughput sequencing; number of samples; forest; karst ecosystem
谷俊锟, 曾馥平, 宋同清, 等. 木论喀斯特常绿落叶阔叶混交林土壤真菌群落多样性特征[J]. 生态科学, 2022, 41(3): 54–61.
GU Junkun, ZENG Fuping, SONG Tongqing, et al. Soil fungal community composition in the Mulun karst evergreen and deciduous broad-leaved mixed forest[J]. Ecological Science, 2022, 41(3): 54–61.
10.14108/j.cnki.1008-8873.2022.03.006
S154.38
A
1008-8873(2022)03-054-08
2020-07-16;
2020-09-08
广西重点研发计划项目(桂科AB17129009); 国家自然科学基金项目(31770495, 31870712, 31971487); 河池市特聘专家项目
谷俊锟(1997—), 男, 湖南张家界人, 硕士, 主要从事生态学研究。E-mail: 505218176@qq.com
杜虎(1986—), 男, 副研究员, 主要从事喀斯特生物多样性研究。E-mail: hudu@isa.ac.cn
猜你喜欢
农业科技与信息(2020年14期)2020-12-18
绿色科技(2019年14期)2019-11-19
乡村地理(2019年2期)2019-11-16
江苏农业科学(2019年5期)2019-09-02
陕西农业科学(2019年6期)2019-07-19
————水溶蚀岩石的奇观
家教世界(2019年4期)2019-02-26
小学生导刊(低年级)(2016年11期)2016-11-14
火控雷达技术(2016年1期)2016-02-06
文化月刊·下旬刊(2014年6期)2014-08-28
数学大王·中高年级(2014年7期)2014-08-06
