
分子筛限域单原子金属催化剂的研究进展
2022-06-29 12:35李加富孙启明
高等学校化学学报 2022年5期
李加富,张 凯,王 宁,孙启明
(1.青岛大学化学化工学院,可持续能源和资源研究院,青岛 266071;2.苏州大学材料与化学化工学部,化学科学国际合作创新中心,苏州 215123)
近年来,负载型单原子金属催化剂由于超高的金属分散度、接近100%的金属利用率以及独特的电子结构被广泛地应用于不同类型的催化反应(如热催化、光催化和电催化等),并展现出优异的催化活性和产物选择性[1~8].目前,不同类型的载体(如碳材料[9,10]、金属氧化物[11~13]、金属有机框架材料(MOFs)[14]、共价有机框架材料(COFs)[15]和分子筛[16,17]等),被用于分散和稳定单原子金属物种.在所有的载体中,分子筛由于具有规则的微孔孔道结构、较大的表面积、可调变的酸碱性和优异的(水)热稳定性被广泛地应用于工业催化、气体吸附/分离和离子交换等领域[18~26],并被认为是限域合成超小尺寸金属物种的理想载体[27~40].如图1所示,在最近的10年来,采用初湿浸渍、离子交换和原位合成等策略,设计和制备了一系列不同类型分子筛(如MFI[16,41~43],FAU[17,44,45],CHA[46],*BEA[47],MWW[48],LTA[49],LTL[50]等)限域贵金属单原子(如Rh[16,41,51~56],Pt[17,47,50,57,58],Ir[44,48,59~62],Au[45,63,64],Pd[17,65],Ru[42,66,67])和非贵金属单原子(如Fe[43,68],Ni[46,69,70],Cu[49,71~73],Ga[74,75],In[76],Ti[77,78])催化剂,并在加氢反应[58]、脱氢反应[47]、氧化反应[41]、异构化反应[17]、水解反应[16]、氨气选择性催化还原氮氧化物[49]及气体吸附/分离领域[70]得到了广泛应用,并展现出优异的反应活性和独特的产物选择性.此外,球差校正-扫描透射电子显微镜(Cs-corrected STEM)、扩展X 射线吸收精细结构(EXAFS)、原位漫反射傅里叶变换红外光谱(DRIFTS)、魔角旋转核磁共振波谱(MAS NMR)和X射线吸收近边结构(XANES)等先进表征技术的不断进步,极大地推动并促进了分子筛限域单原子金属催化剂的研究和发展.分子筛限域单原子金属催化剂的合成与应用已经逐渐成为多相催化领域中重要的研究课题.本文系统地综述了近年来分子筛限域单原子金属催化剂的研究进展,并提出该领域在以后的研究中面临的机遇与挑战.表1 总结了分子筛限域单原子金属催化剂的合成、表征及应用的相关研究结果[16,17,41~43,45~50,53~56,58,60,61,64~66,68,70,71,75~77,79,80].

Fig.1 Schematic illustration of recent advances in zeolite⁃encaged single⁃atom catalysts

Table 1 Summary of the synthesis,characterization and application of zeolite-encaged single-atom catalysts*

Continued
1 分子筛限域单原子贵金属催化剂的合成及应用
1.1 分子筛限域单原子Rh催化剂
20世纪60年代,Rh基配合物催化剂在催化加氢、氢甲酰化、不对称催化等均相催化反应中,得到了广泛的应用[81~83].随后,利用“瓶中造船”、真空吸附等合成策略,多种类型的Rh基金属配合物被限域在大孔分子筛孔道中,制备出一系列具有有机配体保护的单原子Rh 基催化剂[51,52,55,79,84~89].Gates等[51]以乙酰丙酮二(乙烯)铑配合物为金属前驱体,在真空环境下利用物理吸附法,成功地制备了Y型分子筛(FAU 拓扑结构)限域单原子Rh 催化剂.EXAFS 测试表明,所制备的样品中仅存在Rh—C,Rh—O和Rh—Al键,不存在Rh—Rh金属键,证明限域在分子筛内部的Rh物种呈现原子级分布,并通过乙烯配体保护与分子筛骨架上的O 原子键合,稳定于分子筛的孔道中.他们还通过Cs-corrected STEM测试进一步确定分子筛限域Rh物种的单原子特性,如图2(A)所示,基于原子分辨率的原子序数衬度成像技术,在样品的STEM照片中可以清晰地观测到,孤立的Rh原子均匀地分散在FAU分子筛的孔道中[图2(A)中红圈内的白色亮点].所制备的分子筛限域单原子Rh 催化剂被应用于乙烯加氢反应,在室温条件下,具有乙烯配体保护的分子筛限域Rh单原子催化剂并没有乙烯加氢活性.当催化剂在100 ℃氢气还原1 h后,催化剂的活性明显提升,其转化效率(TOF)值可达0.25通过对还原后样品进行热重分析和STEM 测试,发现在升温还原过程中,乙烯基配体逐渐与Rh 原子分离,失去配体保护的相邻Rh原子逐渐聚集形成Rh团簇.相较于Rh单原子,Rh团簇更加有助于反应物H2的吸附解离,进而提升乙烯加氢催化反应活性.
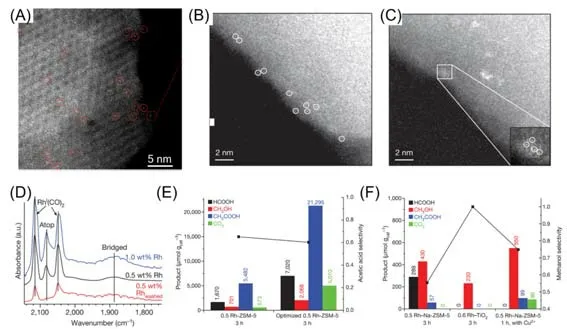
Fig.2 Cs⁃corrected HAADF⁃STEM image of zeolite HY⁃encaged [Rh(C2H4)2]+ catalysts[51](A),Cs⁃corrected HAADF⁃STEM image of as⁃synthesized Rh⁃ZSM⁃5 catalysts with(B) and without(C) washing by water,CO⁃DRIFTS spectra of 1.0%Rh⁃ZSM⁃5,0.5%Rh⁃ZSM⁃5,and 0.5%Rh⁃ZSM⁃5washed(D),catalytic per⁃formance of 0.5% RhZSM⁃5 and optimized 0.5% Rh⁃ZSM⁃5 catalysts in the methane conversion(E),product yields and methanol selectivity for 0.5% Rh⁃Na⁃ZSM⁃5 with and without Cu2+,as well as on 0.6%Rh/TiO2 catalysts(F)[41]
单分散的金属Rh配合物除了可以被锚定在硅铝酸盐分子筛中,还可以被限域在硅磷酸铝(SAPO)分子筛孔道中.近期,Gates等[55]通过在惰性气氛下蒸干有机溶剂的方法,利用SAPO分子筛表面丰富的羟基,通过形成Rh—O—Al 结构,将[Rh(C2H4)2]+离子锚定在具有FAU 拓扑结构的SAPO-37分子筛上.虽然SAPO-37与Y型分子筛具有相同的孔道结构,并且在乙烯加氢反应中展现出相似的活性,但是SAPO-37 负载单原子Rh 催化剂展现出超高的乙烷选择性(约99.6%),而Y 型分子筛负载单原子Rh催化剂却倾向于发生乙烯二聚反应,生成丁烯.上述催化性能的差异可能归因于Y型分子筛负载单原子Rh催化剂中Rh—O键的键长略大于SAPO-37负载单原子Rh催化剂,具有较弱的Rh和O原子间的相互作用以及较强的Brønsted 酸性,更加有利于发生烯烃齐聚反应.除使用乙烯作为配体外,羰基(CO)也常被作为配体保护Rh+离子,用以制备分子筛限域单原子Rh催化剂[84,89].
分子筛除了可以负载单分散的Rh基配合物,还可以作为载体限域无机/有机官能基团保护的单原子Rh 催化剂.Flytzani-Stephanopoulos 等[41]利用“初湿浸渍-纯水洗涤”的合成方法,制备了ZSM-5 分子筛(MFI 结构)负载单原子Rh 催化剂.Cs-corrected STEM 测试表明,通过浸渍合成和水洗处理的Rh-ZSM-5wash样品在550 ℃氢气还原后,Rh 物种呈现单原子分布,并均匀地分散在分子筛的孔道中[图2(B)].而未经水洗处理的Rh-ZSM-5样品在高温还原后,在分子筛的外表面可以明显地观察到尺寸较大的Rh纳米粒子[图2(C)].他们利用原位漫反射红外光谱法,以CO作为探针分子,对所制备的样品进行表征.结果发现,浸渍还原法制备的Rh-ZSM-5样品中不但具有CO分子吸附在Rh单原子上的对称和非对称伸缩振动峰(2116和2049 cm−1),还存在CO分子在Rh纳米粒子上的顶端和桥式吸附特征吸收峰(2082和1885 cm−1),而洗涤后还原的Rh-ZSM-5washed样品中仅存在单原子RhI(CO)2的红外吸收峰[图2(D)].上述测试结果表明,利用纯水洗涤后处理方法可以有效地去除浸渍过程中附着在分子筛外表面的Rh3+离子,避免其在后续高温氢气还原中聚集生成尺寸较大的Rh纳米粒子,从而制备出分子筛限域单原子Rh催化剂.由于单原子Rh超高的金属利用率以及与分子筛上Brønsted酸性位点的协同作用,Rh-ZSM-5wash催化剂在甲烷氧化反应中展现出优异的催化活性,主产物乙酸的选择性超过60%,产率高达21295 μmol/gcat,比含有Rh纳米粒子的Rh-ZSM-5催化剂的性能提升近4倍[图2(E)].他们同时制备了氧化钛负载Rh催化剂(Rh-TiO2)、钠离子交换的Rh-Na-ZSM-5以及采用Cu2+浸渍的Rh-Na-ZSM-5催化剂,上述对比催化剂在甲烷氧化反应中的副产物甲醇选择性分别高达100%,55%和75%,乙酸的产率显著降低[图2(F)].上述催化测试结果表明,分子筛的酸性位点与单原子Rh物种之间的协同作用是甲烷高选择性氧化制备乙酸的关键.
利用初湿浸渍法,Tao 等[54]在真空条件下制备了ZSM-5 负载低金属担载量的单原子Rh 催化剂.EXAFS测试结果表明,具有0.1%金属担载量(质量分数)的Rh/ZSM-5催化剂中仅存在Rh—O键,不含有Rh—Rh金属键,孤立的Rh原子通过氧原子与分子筛骨架上的T原子(Al和Si)连接并稳定存在于分子筛的孔道中[图3(A)].高分辨透射电镜照片中没有观测到明显聚集的Rh纳米颗粒,进一步证实了分子筛中的Rh物种是以单原子形式存在[图3(B)].所制备的Rh/ZSM-5催化剂在甲烷催化氧化反应中的乙酸选择性高达70%.他们利用密度泛函理论(DFT)计算结合13C同位素标记实验,提出了甲烷分子在Rh单原子上氧化生成乙酸的反应路径.如图3(C)所示,反应物CH4和O2分子首先在Rh单原子上活化,并在Rh 单原子上形成—CH3,—OH 和—O 物种;随后反应物CO 分子插入到Rh—OH 中形成—COOH 基团,上述基团进一步与邻近的—CH3基团结合生成第一个乙酸分子,并形成Rh=O 物种;生成的Rh=O可以进一步活化甲烷分子,生成—CH3和—OH基团并吸附在Rh单原子上,最后吸附在Rh原子上的CO分子与相邻的—CH3和—OH基团成键生成第二个乙酸分子.

Fig.3 R⁃space of Rh K⁃edge of experimental(black)and calculated(red)data of the k2⁃weighted Rh K⁃edge EXAFS spectra of spent 0.10%Rh/ZSM⁃5,and the coordination number and bond length of Rh—O,Rh—(O)—Al,and Rh—(O)—Si(A),TEM image of 0.10%Rh/ZSM⁃5(scale bar:10 nm)(B),computa⁃tional studies of reaction pathway of methane oxidation reactions on Rh1O5/ZSM⁃5(C)[54]
分子筛限域单原子Rh催化剂除了可以通过浸渍法制备外,还可以通过原位合成策略制备.近期,于吉红等[16]开发了“配体保护-直接氢气还原”的原位合成策略,以三(乙二胺)三氯化铑(III)配合物和四丙基氢氧化铵为金属前驱体和模板剂,通过一步水热合成法制备了MFI分子筛限域单原子Rh催化剂,合成过程如图4(A)所示.不同于传统的“空气煅烧氢气还原”合成法(易于形成金属团簇或纳米粒子),所制备的分子筛限域Rh配合物样品在不经过煅烧去除有机模板剂的情况下,直接在500 ℃纯氢气条件下进行还原处理.在氢气还原过程中,有机模板剂和配体的氢解与金属的还原同步进行,封装于分子筛孔道中的有机物种,作为保护剂可以有效地抑制Rh原子的聚集,最终制备出分子筛限域单原子Rh 催化剂.EXAFS 测试结果表明,采用直接氢气还原法制备的Rh@S-1-H 催化剂中仅存在Rh—O键,不存在Rh—Rh金属键,证明Rh是以单原子形式锚定在分子筛孔道中[图4(B)].Rh@S-1-H催化剂在CO-DRIFTS 测试中仅存在CO 探针分子吸附在Rh 单原子上的伸缩振动峰(2092 和2023 cm−1),进一步证明限域在分子筛内的Rh物种呈单原子分散状态,并不存在聚集的Rh团簇或纳米粒子[图4(C)].他们还利用Cs-corrected STEM 表征发现Rh 单原子被完全封装在分子筛的晶体内部,并完全被限域在MFI 分子筛十元环交叉孔道中靠近五元环的位置[图4(D)~(F)].由于分子筛的限域作用,所制备的Rh@S-1-H催化剂展现出超高的(水)热稳定性,在700 ℃氢气和氮气氛围下金属Rh依旧可以保持单原子分布,即使在600 ℃水蒸汽中处理5 h 后,金属的平均粒径尺寸仅为1.0 nm.Rh@S-1-H 催化剂在氨硼烷水解产氢反应中展现出优异的催化活性,产氢速率高达432,是采用浸渍法制备分子筛负载Rh催化剂产氢速率的6倍.向分子筛载体中引入Brønsted酸性位点可以进一步提升催化剂的产氢性能,Rh@ZSM-5-H(Si/Al 摩尔比105)催化剂在室温条件下的氨硼烷水解产氢速率可以提升至此外,利用分子筛的孔道择形效应,MFI分子筛限域单原子Rh催化剂在氨硼烷水解制氢串级还原不同分子尺寸的硝基化合物反应中展现出优异的择形催化性能,同时其催化反应速率比使用氢气作为还原剂的反应活性高出3个数量级.

Fig.4 Schematic of the synthetic procedure of the Rh@S⁃1 catalyst(A),Fourier transform of k2⁃weighted EXAFS spectra of Rh foil and various zeolite⁃encaged Rh catalysts at Rh K⁃edge(B), in situ CO⁃DRIFTS spectra of Rh@S⁃1⁃H(Rh single atoms),Rh@S⁃1⁃C(Rh clusters),and Rh@S⁃1⁃H(Rh nanoparticles)(C),Cs⁃corrected STEM images(D,E)of Rh@S⁃1⁃H viewed different orientation as well as the schematic models(F)[16]
1.2 分子筛限域单原子Pt催化剂
近年来,陆续设计制备了系列分子筛限域单原子Pt催化剂,并在烷烃脱氢[17,47]、烯烃/硝基化合物加氢[57,58]和CO 氧化[50,80]等反应中展现出优异的催化性能.李亚栋等[17]利用“配体保护-原位合成”策略,以乙二胺合铂配合物为金属前驱体,一步水热合成法制备出NaY 分子筛限域单原子Pt 催化剂(Pt-ISAS@NaY),其合成过程如图5(A)所示.Cs-corrected STEM 测试表明,单分散的Pt 原子均匀地分布于分子筛晶体内部[图5(B)].EXAFS测试和DFT计算结果表明,Pt-ISAS@NaY 样品中仅存在Pt—O键,不存在Pt—Pt金属键,证明限域在分子筛中的Pt物种呈现单原子分布状态;Pt原子通过和分子筛骨架上的O原子连接,锚定在Y型分子筛的β笼中[图5(C)和(D)].在样品的CO-DRIFTS测试中,仅能观察到CO分子吸附在单原子Pt的特征吸收峰(2166 cm−1),CO脉冲化学吸附测试表明,催化剂中Pt的分散度接近100%,进一步证明了Pt 物种的单原子分散特性.Pt-ISAS@NaY 催化剂在乙烷脱氢反应中展现出优异的催化活性,乙烷转化率为27%,是NaY 分子筛负载的Pt 纳米粒子催化剂Pt NP@NaY 的1.5 倍(金属平均粒径为3.5 nm).此外,Pt-ISAS@NaY 的催化寿命也远高于Pt NPs@NaY,表明单原子Pt催化剂更加有利于抑制乙烷脱氢反应中积碳的生成.此外,酸化后的Pt-ISAS@HY 单原子催化剂在己烷异构化反应中同样具有优异的催化活性,在340 ℃常压氢气反应条件下,Pt-ISAS@HY的转化频率(TOF)高达727 h−1,异构化产物的选择性高达98.1%,其催化活性是相同催化反应条件下Pt NPs@NaY催化剂的5倍.

Fig.5 Schematic of the zeolite⁃encaged Pt single⁃atom catalyst(A),Cs⁃corrected STEM image of Pt⁃ISAS@NaY(B),Fourier transforms of k3⁃weighted Pt L⁃edge EXAFS experimental data for Pt⁃ISAS@NaY as compared with Pt foil and PtO2(C),EXAFS fitting curves of Pt⁃ISAS@NaY at the R space(D)[17]
向Pt 基催化剂中引入非贵金属,不但可以提高贵金属Pt 的分散度,还可以有效提高烷烃脱氢反应过程中烯烃的选择性和催化剂的稳定性.Bell 等[47]利用脱铝的Beta 分子筛(DeAlBEA)为载体,通过浸渍法将Zn2+离子引入到分子筛的缺陷位,形成≡SiOZn—OH 结构;随后,再利用浸渍法将Pt(NH3)4(NO3)2引入到Zn-DeAlBEA 载体上,经空气煅烧处理后最终制备出含有Pt 单原子的Pt-Zn-DeAlBEA 催化剂.EXAFS 测试结果表明,所制备的0.36Zn-DeAlBEA 和0.04Pt-0.36Zn-DeAlBEA样品中Zn—(O)—Si 键的配位数分别为0.8±0.6 和0.5±0.1,Zn—Zn 键的配位数分别为0.8±0.8 和0.6±0.1,证明Zn 物种主要以≡SiOZn—OH 巢穴结构存在,部分Zn 原子位于巢穴的相近位置.Cs-corrected STEM 测试表明,孤立的Pt单原子均匀地分散在分子筛载体上,并锚定在≡SiOZn—OH巢穴中,形成(≡Si—O—Zn)4−6Pt复合结构.0.04Pt-0.36Zn-DeAlBEA催化剂在丙烷脱氢反应中展现出优异的催化性能,在550 ℃条件下,丙烷转化率高达32%,是不含Zn的Pt-DeAlBEA催化剂的13倍,丙烯的选择性大于99%(质量空速为7390 h−1).0.04Pt-0.36Zn-DeAlBEA 催化剂同时具有超高的长效稳定性,经过160 h 的持续稳定性测试后,其丙烷的转化率仅降低10%,当脱氢反应过程中同时引入氢气时,连续反应测试160 h后,催化剂的活性几乎保持不变.
除原位合成法和浸渍合成法外,利用离子交换合成法也可以制备分子筛负载单原子Pt 催化剂.Gates 等[50]以Pt(NH3)4(NO3)2为金属前驱体,采用离子交换法将[Pt(NH3)4]2+离子引入LTL分子筛孔道中,通过在350 ℃氧气氛围下热处理去除NH3配体,制备出LTL分子筛限域Pt单原子催化剂.X射线吸收光谱和高分辨STEM 测试表明,Pt 物种在氧化前后均为原子级分布,位于LTL分子筛十二元环孔道中的Pt 原子在氧化后更倾向于迁移到邻近的八元环孔道中.他们分别测定了氧化前的催化剂和氧化后的PtOx@LTL 催化剂在150 ℃条件下CO 氧化反应活性,结果发现,PtOx@LTL的初始TOF值高达0.012 s−1,是催化活性的3倍以上.此外,随着催化反应时间的延长,催化剂的活性逐渐增强,这归因于在持续氧化反应中Pt物种周围逐渐生成了含氧配体,促进了CO 的氧化反应,进一步说明高分散性的PtOx物种是CO 氧化的活性位点.然而,关于CO 在Pt 单原子上氧化的活性位点仍存在争议.Stair 等[80]利用气相沉法在多级孔HZSM-5分子筛上负载了高分散的Pt物种.通过调变负载于分子筛上的金属Pt担载量,可以调节金属物种中单原子Pt与Pt纳米粒子的比例.通过红外光谱表征和催化测试,他们认为只有Pt纳米粒子具有低温CO氧化活性,由于CO分子对Pt单原子较强的键合能力,导致Pt单原子在CO氧化反应中表现为惰性.
分子筛负载单原子Pt 催化剂不但可以应用于烷烃脱氢、烷烃异构化以及CO 氧化等反应,还在加氢还原反应中具有较高的活性.李兰冬等[58]以氯酸铂和3-(2-氨基乙基氨基)丙基三甲氧基硅烷为金属前驱体和配体,利用一步水热晶化法成功地制备了Y 型分子筛限域单原子Pt 催化剂(Pt@Y).Cs-corrected STEM 测试表明,Pt 单原子均匀地分散于分子筛的晶体内部;通过对Y 型分子筛的孔道进行理论模拟,他们确定Pt单原子位于方钠石笼和FAU超笼共享的六元环中.EXAFS和XANES测试结果进一步确定,限域在分子筛内的Pt 物种以单原子Pt 形式存在,并与分子筛骨架上的O 原子相互作用,带有部分正电荷(Ptδ+).Pt单原子和分子筛形成的Ptδ+···O2−结构有助于将氢气异裂生成H+和Hδ-物种,而非中性的H物种.这种带有电荷的H物种在催化α,β-不饱和醛类化合物加氢反应中,可以显著地提高碳氧双键加氢的选择性,并同时降低碳碳双键的加氢活性,最终生成不饱和醇的选择性超过86%.相比之下,利用浸渍法制备的分子筛负载Pt纳米粒子催化剂在相同条件下,不饱和醇的选择性低于50%.此外,Pt@Y在芳香族硝基化合物还原反应中也展现出优异的催化活性,在底物完全转化的情况下,对应的氨基化合物选择性均在89%以上.
1.3 分子筛限域单原子Ir催化剂
在目前绝大多数报道中,分子筛限域单原子Ir 催化剂通常要使用羰基或乙烯配体来稳定Ir 单原子[44,59~61,89~92].如,Gates等[90]利用真空吸附乙酰丙酮双(乙烯)铱(I)配合物的方法,在脱铝的Y型分子筛上制备了负载量为1%(质量分数)的单原子Ir催化剂.EXAFS测试结果显示,催化剂中不存在Ir—Ir金属键,表明限域在分子筛内的Ir 物种是以单原子形式存在;每个Ir 原子与4 个碳原子相连(键长为0.21 nm),表明制备过程中乙烯配体保持稳定.此外,每个Ir 原子中还存在2 个键长为0.215 nm 的Ir—O 键,证明催化剂中Ir(C2H4)2配合物通过与分子筛骨架上的O 原子连接,稳定在分子筛的孔道之中.所制备的分子筛限域单原子Ir催化剂在乙烯加氢的反应中具有催化活性,在室温条件下,催化剂的初始乙烯转化率达到20%,稳态条件下的TOF值为0.07当连续反应10 h后,反应后的催化剂仍然没有观测到明显的Ir—Ir金属键,证明催化剂中的Ir物种一直保持原子级分布;反应后的样品中Ir—C 键配位数逐渐减少,这可能是由于在反应过程中生成了新的反应中间体,替代了原有的乙烯配体或者覆盖在了Ir 原子的表面,从而降低了Ir 的反应活性.他们进一步利用Cs-corrected STEM 测试,成功地解析了催化剂中Ir 原子在分子筛孔道中的位置,并提出在热处理情况下,单原子Ir聚集形成纳米团簇的机制[44].如图6(A)和(B)所示,单原子Ir均匀地分散于分子筛的孔道之中,通过对图像进行傅里叶滤波图像处理并结合FAU分子筛结构模拟,他们最终确定Ir原子位于分子筛骨架铝中心附近的2种位置:即三空位(T5)和六元环中心处(T6).上述判断与催化剂的合成过程中,Ir+(C2H4)2易于吸附到分子筛酸性羟基位点相符.此外,样品经过120 ℃氢气还原处理后,部分Ir单原子聚集形成了Ir4和Ir6两种类型的金属团簇.通过对金属原子以及团簇的位置统计,他们发现Ir单原子和团簇都更倾向于存留于FAU分子筛的T6位置,这可能是由于在还原过程中,锚定在T5位置的Ir原子上的配体容易发生脱离,Ir原子更愿意迁移到T6位置,最终聚集形成Ir金属团簇.

Fig.6 Cs⁃corrected HAADF⁃STEM images and the corresponding models illustrating the positions of the Ir+ ions for T6 site and the T5 site(A,B)[44],HAADF⁃STEM images of Ir@MWW⁃560⁃air sample along different orientations(C,D),model of MWW zeolite with isolated Ir atoms located at different positions and simulated HAADF⁃STEM image(E),in situ EXAFS spectra of 0.24Ir@MWW samples reduced at given temperature by H2(F)[48]
虽然向分子筛限域单原子Ir催化剂中引入配体可以对金属起到稳定作用,但是在高温、高压或氧化还原等催化反应条件下,配体容易从催化剂中脱离,导致金属容易发生烧结团聚.制备高稳定性且无配体保护的分子筛限域单原子Ir催化剂仍具有一定挑战.近期,Corma等[48]使用乙二胺合铱配合物和N,N,N-三甲基-1-金刚烷基氢氧化铵作为金属前体和有机模板剂,通过一步水热合成法制备了MWW分子筛限域单原子Ir催化剂Ir@MWW-air(样品在560 ℃空气中煅烧去除有机模板剂和配体),Ir的担载量约为0.24%.Cs-corrected STEM 测试结果表明,在分子筛的外表面没有明显的金属颗粒的团聚,孤立分散的Ir原子被完全封装在分子筛的孔道内,并位于连接两个相邻的十二元环超笼的十元环窗口处[图6(C)~(E)].EXAFS测试结果显示,样品中只存在Ir—O键,并不存在Ir—Ir金属键,进一步证实了Ir物种的单分散特性[图6(F)].当样品经过氢气还原处理后,在EXAFS谱图中可以明显观测到形成Ir—Ir金属键,随着还原温度的逐渐升高,Ir—Ir金属键的平均配位数也逐渐增大,而Ir—O键的平均配位数逐渐减少,这表明在高温氢气氛围下,单分散氧化态的IrOx物种逐渐被还原为亚纳米Ir金属团簇.CO-DRIFTS 测试结果表明,当还原温度从400 ℃升高至650 ℃时,CO与金属Ir配位的特征吸收峰由2020 cm−1逐步蓝移至2065 cm−1,证明Ir物种的粒径随着还原温度的升高而逐渐增大.虽然限域在分子筛孔道内的金属在高温还原后金属尺寸有所增大,但是平均金属团簇粒径依旧小于1 nm,呈亚纳米级别分布.与之对比,利用浸渍法制备的Ir/MWW-imp催化剂,在经过650 ℃还原后,大量的金属颗粒(粒径大于5 nm)聚集在分子筛外表面,这表明采用原位合成的策略所制备的分子筛限域金属催化剂具有更高的热稳定性.原位合成的Ir@MWW-500H2催化剂在丙烷氢解反应展现出优异的催化活性,在相同催化条件下,Ir@MWW-500H2的催化活性是浸渍法制备的Ir/MWW-imp-500H2催化剂的17倍.
1.4 分子筛限域其它贵金属单原子催化剂
分子筛除了可以限域合成Rh,Pt和Ir单原子催化剂外,还可以制备其它贵金属单原子催化剂,如Au[45,63,64],Pd[65]和Ru[42,66,67]等.Gates等[63]以二甲基(乙酰丙酮)金配合物为金属前驱体,采用真空物理吸附法,制备了NaY 型分子筛封装单原子Au 催化剂.EXAFS 测试结果表明,催化剂只存在Au—O 键(键长0.221 nm)、Au—C键(键长0.192 nm)和Au—Al键(键长0.326 nm),不存在Au—Au金属键.限域在分子筛孔道中的Au 物种是以配合物的形式存在,并通过O 原子与分子筛骨架上的Al 原子连接.在后续的研究中,他们利用Cs-corrected STEM测试进一步证实Au物种的单原子分布状态,并且在分子筛表面不存在Au团簇[图7(A)],与EXAFS测试结果相符[45].结合DFT模拟计算,确定Au单原子位于FAU超笼中靠近铝中心的T5和T6位置[图7(B)].分子筛限域单原子Au催化剂在室温下催化CO氧化的反应速率可达0.026但是反应15 min后,催化剂活性降低至0.0029 molCO·s−1·EXAFS测试表明,反应后的样品中依旧没有Au—Au金属键的生成,在Cs-corrected STEM照片中也捕捉不到Au 团簇的生成,说明催化剂中的Au 物种在反应过程中一直保持原子级分散.通过对样品的STEM照片中Au原子位置的统计发现,随着反应进行,位于T6位点的Au原子占比由反应前的34%增加到73%,说明Au原子在CO氧化过程中在分子筛的孔道中发生位置的迁移.同时,通过CO-DRIFTS和XANES测试发现,催化剂中的乙酰丙酮配体随着反应的进行,逐渐从配合物中脱离,Au(III)物种也被还原为Au(I),上述变化可能是导致催化剂反应活性迅速下降的原因.

Fig.7 Cs⁃corrected HAADF⁃STEM images of the sample prepared by adsorption of Au(CH3)2(acac)in zeo⁃lite NaY(A),perspective views of gold atoms at T6 and T5 positions in an isolated FAU supercage,viewed from the[110]and[111]projections(B)[45],TEM images(C)of 0.01%Pd/ZSM⁃5 and 2.0%Pd/ZSM⁃5 catalysts,Fourier transform magnitudes of k2⁃weighted EXAFS data of 0.04%Pd/ZSM⁃5,Pd foil,and PdO nanoparticles(D)[65] schematic of synthesis procedure of Ru SAs/S⁃1(E),Cs⁃corrected HAADF⁃STEM image of Ru SAs/S⁃1(F)[42]
Flytzani-Stephanopoulos 等[64]采用初湿浸渍法在将KAu(CN)2金属前驱体引入到KLTL 分子筛孔道中,并通过固相浸渍法引入大量的K+离子来稳定Au 原子,在煅烧除去氰基配体后,制备了单原子Au催化剂[Au-K/KLTL,Au的担载量(质量分数)为0.25%].通过向催化剂中引入K+离子,可以显著抑制Au物种的聚集,Au的分散度甚至可以达到100%,呈原子级分布.STEM测试表明,分子筛的外表面不存在Au 纳米粒子,样品的EXAFS 谱图中也存在Au—O 键(键长为0.208 nm)和Au—K 键(键长为0.343 nm),进一步说明K+离子易于诱导高分散的金原子形成.Au-K/KLTL 催化剂在水煤气变换反应中展现出显著提升的催化性能,在150 ℃条件下,其催化反应速率可达,是不引入K+离子制备的Au/KLTL催化剂催化性能的8倍以上.
Tao 等[65]利用真空吸附-初始浸渍合成策略,制备了ZSM-5 分子筛负载超低金属担载量的单原子Pd催化剂[Pd担载量(质量分数)为0.01%~0.04%].如图7(C)所示,在样品的透射电子显微镜照片中没有观察到明显聚集的金属纳米粒子,而使用相同合成方法制备的担载量为2%的样品中却存在着明显的金属纳米粒子,说明低的担载量是制备单原子Pd催化剂的关键.EXAFS测试表明,Pd/ZSM-5中仅存在键长为0.2 nm的Pd—O键(配位数约为4),不存在Pd—Pd金属键,证明限域在分子筛中的Pd物种是以单分散的Pd1O4形式存在[图7(D)].担载量为0.01%的Pd/ZSM-5催化剂展现出催化过氧化氢氧化甲烷的反应性能,在95 ℃,3×10−5Pa 反应条件下,甲烷的转化率可达12.3%,含氧产物总收率可达234 μmol,但是产物中的甲醇占比很低,主要产物是甲酸.为了提高甲醇收率,他们利用双浸渍合成方法,制备了含有0.01%Pd和2%CuO的分子筛限域双金属催化剂.在反应过程中,CuO物种可以促进分解多余的过氧化氢,抑制甲醇被进一步氧化生成甲酸,最终甲醇的选择性高达86.35%,生成速率高达2.33
除了浸渍合成法外,原位合成策略也被应用于制备分子筛限域单原子金属催化剂.姜久兴等[42]利用“配体保护-原位合成”策略,以乙二胺合钌配合物作为Ru的前驱体,一步水热合成法制备了MFI分子筛限域单原子Ru催化剂,其合成步骤如图7(E)所示.为了实现金属Ru物种在分子筛中呈原子级分布,他们采用了真空热解的方式去除分子筛孔道中的有机模板剂和配体.Cs-corrected STEM测试表明,高分散的Ru原子均匀地分布于MFI分子筛晶体之中,在分子筛的外表面没有发现明显的金属团簇聚集[图7(F)].EXAFS 测试结果表明,催化剂只存在键长为0.201 nm 的Ru—O 键,不存在键长为0.268 nm的Ru—Ru金属键,进一步证实了Ru物种的单原子分散特性.该分子筛限域单原子Ru催化剂被应用于热催化合成氨反应,在375 ℃和常压反应条件下,氨气生成速率为6.1,并且反应100 h后催化剂的活性没有明显降低.当向催化剂中引入Ba2+离子作为助催化剂时,在400 ℃和常压条件下,合成氨的反应速率高达1389.5
2 分子筛限域非贵金属单原子催化剂的合成及应用
2.1 分子筛限域单原子Fe催化剂
近年来,分子筛负载单原子Fe催化剂在甲烷氧化制甲醇[68]、烃类物质燃烧[93]、烷烃脱氢[43]及氨气选择性还原[93~95]等反应中展现出优异的催化性能.Solomon 等[68]以乙酰丙酮铁作为金属前驱体,采用浸渍法制备了Beta分子筛负载单原子Fe催化剂.通过圆二色光谱及穆斯堡尔谱测试和DFT理论模拟计算,他们发现催化剂中的Fe以单分散的Fe(II)形式存在,并被限域在Beta分子筛六元环的中心.所制备的分子筛负载单原子Fe催化剂在甲烷氧化制甲醇反应中具有优异的催化活性,在室温条件下,甲醇的产率可达70%~80%.
肖丰收等[43]以乙二胺四乙酸合铁作为金属前驱体,利用原位水热合成的策略,制备了纯硅Silicalite-1分子筛负载单原子Fe催化剂(FeS-1-EDTA),金属Fe的担载量(质量分数)为0.8%.STEM测试表明,在分子筛的外表面以及内部均不能观测到明显团聚的金属物种,金属Fe物种以单原子的形式存在于分子筛的晶体之中[图8(A)和(B)].在样品的紫外拉曼光谱中,仅存在归属于Feδ+—O—Si的对称和非对称伸缩振动特征峰(515和1155 cm−1),不存在分子筛骨架外Fe物种的特征峰,证明Fe是以孤立的单原子Fe 位点稳定于分子筛的骨架中[图8(C)].EXAFS 测试表明,FeS-1-EDTA 之中只存在Fe—O 键,不存在Fe—Fe 金属键,进一步证实了Fe 的单原子分散特性[图8(D)].FeS-1-EDTA 催化剂在乙烷脱氢反应中展现出了优异的催化性能,在600°C 常压催化条件下,乙烷的转化率可达26.3%,乙烯的选择性高达97.5%,高于目前报道的其它类型的非贵金属催化剂和贵金属催化剂[图8(E)].此外,FeS-1-EDTA 催化剂在乙烷脱氢反应中还具有超高的稳定性,在持续反应200 h后,催化剂的反应活性几乎保持不变,EXAFS和紫外拉曼光谱测试证明,在反应过程中,催化剂中金属物种的结构和性质没有发生改变,Fe物种一直保持单原子分布.

Fig.8 Cs⁃corrected HAADF⁃STEM images of FeS⁃1⁃EDTA with different amplifications(A,B),UV resonance Raman(λex=325 nm)(C)and EXAFS spectra of fresh and spent FeS⁃1⁃EDTA catalysts after the EDH re⁃action(D),ethane conversion and ethylene selectivity of various catalysts in EDH reactions(E)[43]
2.2 分子筛限域单原子Ni催化剂

Fig.9 Schematic structure evolution of nickel species confined in CHA zeolite(A),comparison of different catalysts in acetylene⁃selective hydrogenation(B) and calculated Gibbs free energy profile of Ni@CHA⁃catalyzed acetylene⁃selective hydrogenation at 453 K(C)[46]
分子筛限域单原子Ni 催化剂近年来也不断被报道,并在炔烃选择性加氢[46,69]、炔烃与烯烃的分离[70]领域展现出优异的性能.李兰冬等[46]以二乙烯三胺合镍配合物为模板剂和金属前驱体,利用原位水热合成策略制备了CHA分子筛限域单原子Ni催化剂[图9(A)].在未煅烧样品的紫外-可见-近红外(UV-Vis-NIR)漫反射谱图中,可以看到六配位镍的特征吸收峰,说明二乙烯三胺合镍配合物在分子筛的合成过程中,结构保持完好.经过高温空气煅烧后,镍配合物中的有机配体被逐渐去除,Ni物种与分子筛骨架上的氧原子配位,锚定在分子筛的孔道中.EXAFS测试结果显示,煅烧后的Ni@CHA样品中只存在Ni—O键(平均配位数为4)和Ni—Si(Al)键(平均配位数为1.9),不存在Ni—Ni金属键,证明分子筛中的Ni 物种是以单原子形式限域在CHA 分子筛的六元环内.Na+离子交换后制备的Na-Ni@CHA 催化剂在乙炔选择性加氢制乙烯反应中展现出优异的产物选择性,当乙炔转化率达到100%时,乙烯的选择性超过97%,远高于浸渍法制备的NiO/Na-CHA(选择性<60%)以及离子交换法制备的Ni-CHA(选择性<80%),也超过了常用的Pd-Ag/Al2O3催化剂(乙烯选择性约90%)以及商用林德拉催化剂Pd-PbO/CaCO3(约75%)[图9(B)].向催化剂中引入不同种类的碱金属离子(如Li+,Na+和K+),可以改变乙炔加氢反应性能,随着碱金属离子尺寸的不断增大,催化剂的乙烯选择性也随之提高,但过大的碱金属离子尺寸同时会带来位阻效应,降低了乙炔转化率.此外,Na-Ni@CHA催化剂在乙炔加氢中具有较好的稳定性,在20 h的连续反应后,催化性能仍能保持稳定.在模拟工业乙烯进料(乙烯占比50%,乙炔占比0.5%)的催化性能测试中,Na-Ni@CHA催化剂仍能够保持较好的催化活性,乙烯选择性仍可以达到90%左右.利用原位红外表征和DFT计算,他们提出了在Ni@CHA单原子催化剂上乙炔加氢的反应路径:如图9(C)所示,H2分子首先在Ni@CHA催化剂上发生解离,一个H原子吸附在单原子Ni上形成N—H键,另一个H原子吸附于分子筛骨架的氧原子上形成质子H.在随后的加氢反应过程中,Ni—H 与乙炔反应生成Ni—C2H3的能垒小于Ni—H 与乙烯反应生成Ni—C2H5的能垒,同时Ni—C2H3中间体与骨架上的质子氢反应生成乙烯的能垒,也远低于Ni—C2H5中间体与质子氢反应生成乙烷的能垒.因而,Ni@CHA催化剂在乙炔加氢反应中,更易于生成乙烯而非过度加氢得到的乙烷.
除了八元环小孔分子筛,具有十二元环的大孔分子筛也可以作为载体限域单原子Ni催化剂.李兰冬等[70]通过向分子筛合成体系中引入二乙烯三胺基丙基三甲氧基硅烷,成功地制备了FAU分子筛限域单原子Ni 催化剂(Ni@FAU).同步辐射X射线粉末衍射、透射电子显微镜和X射线光电子能谱测试结果表明,Ni@FAU中不存在聚集的金属粒子,Ni物种以单分散的Ni(II)形式存在.通过原位中子粉末衍射及DFT计算,他们确认单分散的Ni(II)位点稳定于FAU分子筛的六元环孔道处.所制备的分子筛限域单原子Ni催化剂在炔烃/烯烃的选择性分离中展现出优异的性能,当乙炔分压为2.026×105Pa时,乙炔的吸附量高达2.0 mmol/g,远高于乙烯的吸附量.在程序升温脱附测试中,样品的乙炔吸附量以及脱附温度均显著高于乙烯,证明Ni@FAU 对于乙炔具有很好的吸附选择性.在乙炔/乙烯的穿透测试中,Ni@FAU 的乙炔吸附量为1.72 mmol/g,高于同类型的Zn@FAU(0.91 mmol/g)和Cu@FAU(1.26 mmol/g),以及JCM-1,UTSA-200a 等有机金属框架材料.而Ni@FAU 的乙烯吸附量低至0.02 mmol/g,乙炔/乙烯的吸附选择性接近100%,远高于其它负载型金属材料.此外,Ni@FAU 在丙炔/丙烯、丁炔/1,3-丁二烯的分离中也展现出优异的性能,炔烃的动态分离选择性分别高达92%和83%.
2.3 分子筛限域其它非贵金属单原子催化剂
除Fe和Ni以外,近年来,其它类型非贵金属单原子(如Cu[71~73],Ga[74,75],In[76],Ti[77,78,96,97]等)也成功地被限域在分子筛的孔道中.Ribeiro等[98]使用H-SSZ-13(CHA拓扑结构)分子筛作为载体,通过离子交换的方法制备了分子筛限域单原子Cu 催化剂(Cu-SSZ-13).在Cu-SSZ-13 催化剂的EXAFS 谱图中,只有键长为0.194 nm的Cu—O键(配位数为4)和Cu—(O)—Si键,不存在Cu—Cu金属键,结合DFT计算,他们确定孤立的Cu离子位于CHA分子筛六元环的中心.Cu-SSZ-13催化剂在氮氧化物选择性还原反应中的速率最高可达3.8×10−6.他们还通过原位X 射线吸收光谱并结合DFT 理论计算,阐述了在氮氧化物选择性还原反应中,单分散的Cu(I)/H+和Cu(II)形式之间的闭合氧化还原循环[图10(A)][71].除了CHA 分子筛外,LTA 分子筛也被用于限域合成单原子Cu 催化剂.Hong 等[49]利用离子交换法,成功制备高硅铝比的LTA 分子筛限域单原子Cu 催化剂(Cu-LTA).电子自旋共振(ESR)光谱以及同步辐射XRD测试测试结果表明,高度分散的Cu(II)离子位于LTA分子筛六元环的中心.所制备的单原子Cu-LTA 催化剂在NH3选择性还原NOx反应中具有超高的热稳定性,在900 ℃蒸汽老化后,催化剂仍然能保持优异的脱硝活性,NO 转化率仍能够保持在90%;而经过蒸汽老化处理的Cu-SSZ-13催化剂的NO转化率却低于50%.此外,在SO2中毒失活再生测试中,再生后的Cu-LTA催化剂能够完全恢复初始催化活性,而再生后的Cu-SSZ-13催化剂活性却下降到原来的80%以下.
Bell 等[74]以GaCl3为金属前驱体,通过气相交换法制备了MFI 分子筛限域单原子Ga 催化剂.XANES以及EXAFS测试结果表明,限域在分子筛孔道中的Ga物种的化合价为+3价,并且与分子筛骨架上的4个O原子配位.当Ga/Al 摩尔比≤0.3时,催化剂的拉曼光谱中不能检测到Ga2O3物种的存在,进一步证明Ga物种的单原子分散特性.通过红外光谱以及氨气程序升温脱附测试,证实Ga物种主要以[Ga(OH)2]+-H+的形式存在于分子筛的孔道中.样品进行氢气还原后,Ga 物种的存在形式变为[Ga(OH)H]+-H+,[GaH2]+-H+和[GaH]2+3 种形式.所制备的MFI 分子筛限域单原子Ga 催化剂在丙烷脱氢反应中具有活性,通过反应动力学以及DFT计算研究发现,[GaH]2+阳离子是丙烷脱氢的催化活性中心[75].除Ga 外,同属于第三主族的In 也可以以单原子分散的形式限域在分子筛孔道中.Shimizu等[76]采用固态离子交换法,制备CHA分子筛限域单原子In催化剂.如图10(B)所示,催化剂的EXAFS谱图中只有In—O键,观察不到In—In键的存在,证明In物种以单原子形式存在.催化剂在乙烷脱氢反应中展现出优异的催化性能,乙烷的转化率和乙烯的选择性分别高达25.9%和96.1%,催化寿命可达90 h以上.基于反应动力学、原位红外光谱和DFT计算结果,证明[InH2]+阳离子是乙烷脱氢的催化活性位点.

Fig.10 Proposed NH3⁃SCR cycle over Cu⁃SSZ⁃13 catalyst[71](A),EXAFS spectra of In⁃CHA after reductive solid⁃state ion⁃exchange under H2 atmosphere at room temperature(B)[76],schematic representation of zeolite supported calix[4]arene⁃TiIV(C)[77],grafting model of calix[4]arene⁃TiIV in the confining 12⁃MR ring(D)[78]
Katz等[77,78]以杯[4]芳烃钛配合物为金属前驱体,通过配合物与分子筛硅羟基的缩合反应,制备了UCB-4分子筛限域单原子Ti催化剂[图10(C)].单分散的钛物种主要位于末端T原子位置处和表层的十二元环口袋内.环己烯环氧化反应的测试表明,当钛物种与末端T原子松散地连接在一起时,反应的速率常数为(9±2)L2·mol−2·s−1,而当钛物种被限制在十二元环的口袋内[图10(D)],速率常数提高到(48±8)L2·mol−2·s−1,说明分子筛限域作用可以提升催化反应活性.
3 总结与展望
分子筛限域单原子金属催化剂在近年来得到了研究者们的广泛关注,并在多相催化反应中展现出优异的催化活性和特定产物的选择性.重要的是,与其它负载型单原子金属催化剂相比,分子筛限域单原子金属催化剂具有更加优异的(水)热稳定性,同时分子筛规则的孔道结构还赋予该类催化剂独特的择形催化性能.此外,分子筛固有的酸性和碱性位点可以与金属单原子产生协同效应,进一步提升催化剂的反应活性,甚至创造新的反应路径.分子筛限域单原子金属催化剂的发展极大地促进了多相催化领域的进步.在未来的研究过程中,分子筛限域单原子金属催化剂的合成与应用领域仍然存在较大的发展空间,但是在某些方面也面临一定挑战.
发展更多新颖、简单且绿色的合成策略制备分子筛限域单原子金属催化剂,是推动该领域向前发展的最大动力.目前,分子筛限域单原子金属催化剂的合成策略主要有两种:(1)分步合成策略,即先合成分子筛载体,而后通过浸渍法或离子交换法将原子级分散的金属原子锚定在分子筛载体上;(2)原位合成策略,即在合成分子筛的母液中直接引入金属物种,金属物种随着分子筛的晶化过程被负载在分子筛载体上.浸渍法是最常用的制备分子筛负载单原子金属催化剂的方法,但是采用上述方法所制备的大部分金属物种并没有被限域到分子筛的孔道内,而是附着在分子筛晶体的外表面.在后续的高温还原过程或实际催化反应中,金属单原子极易发生烧结团聚,导致性能下降.离子交换法虽然有利于金属物种在分子筛载体上呈现原子级分布,但是该方法只能用于具有离子交换能力的硅铝酸盐分子筛载体,对于中性的纯硅分子筛并不适用.相比而言,通过原位合成策略制备的金属单原子更容易被完全封装在分子筛晶体的内部,展现出更加优异的稳定性和择形催化性能.然而截至目前,原位合成策略仅在MFI,FAU和MWW等少部分类型分子筛中得以成功应用,进一步发展原位合成策略,使不同种类的金属单原子可以被完全封装在更多拓扑结构的分子筛晶体内部仍具有挑战.优化合成条件(如晶化温度、晶化时间及合成配比等),并选择合适的分子筛合成原料以及适合的金属前驱体是实现原位合成分子筛限域单原子金属催化剂的关键.
此外,为了保证金属物种的原子级分散,原位合成的分子筛限域单原子金属催化剂(尤其是贵金属)中的金属担载量(质量分数)往往低于1%.优化原位合成体系中金属物种(尤其是金属配合物)的尺寸和构型与分子筛孔道结构的匹配性,并设计构筑更多可以锚定金属物种的分子筛缺陷位点可能是提高金属负载量的途径.目前,分子筛限域的金属单原子可以分为贵金属单原子和非贵金属单原子.通常情况下,非贵金属比贵金属更加活泼,表面能更高,因而理论上非贵金属以原子级单分散形式稳定于分子筛载体上更加困难.但是,部分非贵金属往往更容易通过氧原子与分子筛骨架上的硅原子或者铝原子相连,并利用分子筛特定的孔道结构(如五元环、六元环等)的限域作用,反而比贵金属更加容易稳定存在于分子筛的孔道之中.因此,选择分子筛限域单原子金属催化剂的合成策略时,还需要同时考虑分子筛载体的结构特性和金属物种的种类等因素.
目前,可以通过Cs-corrected STEM,CO-DRIFTS 和EXAFS 等表征技术确定催化剂中金属物种的单原子分布特性,但是精准确定金属原子在分子筛(尤其是具有三维结构的分子筛)孔道中的位置仍然具有挑战.近年来,积分差分相位衬度扫描透射电子显微技术(iDPC-STEM)的迅速发展为确定限域在分子筛内部的金属单原子的位置和结构信息提供了便利和更为直观的证据[34,99].但是值得注意的是,上述表征单原子金属物种的手段均依赖于使用比较昂贵的测试仪器,测试资源有限.因而,亟待开发更多便捷、廉价且高效的测试手段方便快速地确定所制备的材料中单原子金属特性.此外,虽然单原子催化剂在某些催化反应中展现出优异且独特的催化性能,但是单原子催化剂在部分反应中的催化机制仍不清晰甚至存在争议.在很多情况下,负载在分子筛上的单原子物种的结构会随着反应推进不断发生变化,有些金属单原子在反应过程中会聚集生成尺寸较大的金属团簇或纳米颗粒,这为准确分析和指认催化剂的活性位点带来困难.发展先进的原位表征技术(如原位Cs-corrected STEM、原位红外、原位XAFS等),实现对催化剂在工况反应条件下的原位表征,可以使研究者更直观地观测到催化剂在反应过程中活性位点的实时结构变化,从而更好地研究和理解分子筛限域单原子金属催化剂在催化反应中的构效关系和催化机制.
猜你喜欢
内燃机与动力装置(2022年1期)2022-03-21
煤气与热力(2021年9期)2021-11-06
陶瓷学报(2021年2期)2021-07-21
哈尔滨工程大学学报(2021年2期)2021-03-19
中国造纸(2020年7期)2020-08-11
湖北农机化(2020年4期)2020-07-24
无机化学学报(2020年7期)2020-07-20
筑路机械与施工机械化(2017年5期)2017-08-31
科技创新导报(2016年30期)2017-03-15
郑州大学学报(工学版)(2014年6期)2014-03-01
