
超高效液相色谱-串联质谱法同时测定动物源性食品中46 种食源性兴奋剂残留量
2022-08-02 03:11张海超张婧雯艾连峰黄雪静
食品科学 2022年14期
张海超,王 敬,洪 灯,张婧雯,田 浩,*,艾连峰,黄雪静
(1.石家庄海关技术中心,河北 石家庄 050051;2.杭州海关技术中心,浙江 杭州 310000)
近年来在畜牧养殖生产中,由于各种饲料添加剂和人工激素的广泛使用导致畜禽肉制品中产生兽药残留的现象屡见不鲜。每年国家食品安全监督抽查中β-受体激动剂、糖皮质激素、蛋白同化剂等药物残留常被检出,也成为动物源性食品的重点监控项目[1]。残留的这些药物也给体育赛事造成食源性兴奋剂的困扰[2]。我国发布了《冬奥会食品安全食品动物食源性兴奋剂类药品控制管理规范》,该规范包含40余项动物食源性兴奋剂,有激动剂、蛋白同化剂、糖皮质激素和利尿剂等。针对每一种或每一类化合物进行分析耗时较长、浪费人力物力,因此,研究多种食源性兴奋剂的快速筛查方法对于检测效率、快速准确出具监测数据显得尤为重要。
目前,针对食品中这些兴奋剂的检测方法有气相色谱-质谱[3]、液相色谱[4-6]、液相色谱-串联质谱(liquid chromatography-tandem mass spectrometry,LC-MS/MS)[7-16]和液相色谱-高分辨质谱[17-19]等。但针对这些化合物同时检测的技术研究却很少,难以满足重大体育赛事对于时效性的要求。齐鹤鸣等[20]采用LC-MS/MS测定牛肉中18 种兴奋剂残留,包括β-受体激动剂、类固醇激素、糖皮质激素和玉米赤霉醇。该方法虽然针对多种食源性兴奋剂进行检测,但是覆盖面较窄,无法满足冬奥会监管项目要求。马俊美等[21]采用四极杆/飞行时间质谱测定牛肉中16 种蛋白同化制剂、8 种利尿剂、6 种糖皮质激素共30 种食源性兴奋剂。该方法简便、快速,但四极杆/飞行时间质谱这种高分辨质谱价格昂贵,大部分实验室仍未普及,LC-MS/MS仪仍是当前检测机构的主流检测手段,现行有效检测标准方法也大多采用LC-MS/MS法[22-24]。且由于动物源性食品含有较多的蛋白、脂肪和磷脂,为保证数据的准确性,减少对色谱柱和仪器的污染,需要对其进行净化处理。近年来在多种兽药快速分析中多采用PRiME HLB固相萃取柱[25-28]和QuEChERS法[29-32]净化。为使方法普适性更强,能够为大多数实验室使用,本研究针对冬奥会指定检测项目对PRiME HLB固相萃取和QuEChERS法的净化效果进行比较,采用LC-MS/MS仪建立动物源性品中10 种β-受体激动剂、3 种β-受体阻断剂、13 种蛋白同化剂、9 种糖皮质激素、11 种利尿剂共计46 种食源性兴奋剂的检测技术。方法操作简便、快速,为多种食源性兴奋剂的测定提供了技术参考。
1 材料与方法
1.1 材料与试剂
5 种动物源食品样品(猪肉、牛肉、羊肉、鸡蛋和牛奶)随机购买于河北石家庄当地农贸市场。乙腈、甲醇(均为色谱纯) 德国Merck公司;甲酸(色谱纯)德国Fluka公司;乙酸、乙酸钠、氯化钠(均为分析纯)广州化学试剂厂;高纯水 美国Millipore公司。
克伦特罗(CAS号:37148-27-9)、沙丁胺醇(CAS号:18559-94-9)、莱克多巴胺(CAS号:97825-25-7)、特布他林(CAS号:23031-25-6)、氯丙那林(CAS号:3811-25-4)、非诺特罗(CAS号:13392-18-2)、妥布特罗(CAS号:41570-61-0)、喷布特罗(CAS号:36507-48-9)、西马特罗(CAS号:54239-37-1)、普萘洛尔(CAS号:525-66-6)、阿替洛尔(CAS号:29122-68-7)、美托洛尔(CAS号:37350-58-6)、克伦丙罗(CAS号:38339-11-6)、美雄酮(CAS号:72-63-9)、司坦唑醇(CAS号:10418-03-8)、甲基睾酮(CAS号:58-18-4)、丙酸睾酮(CAS号:57-85-2)、诺龙(CAS号:434-22-0)、丙酸诺龙(CAS号:7207-92-3)、苯丙酸诺龙(CAS号:62-90-8)、勃地酮(CAS号:846-48-0)、群勃龙(CAS号:10161-33-8)、睾酮(CAS号:58-22-0)、去氢表睾酮(CAS号:53-43-0)、泽伦诺(CAS号:26538-44-3)、齐帕特罗(CAS号:117827-79-9)、泼尼松(CAS号:53-03-2)、泼尼松龙(CAS号:50-24-8)、地塞米松(CAS号: 50-02-2)、倍他米松(CAS号:378-44-9)、氟氢可的松(CAS号:37148-27-9)、甲基泼尼松龙(CAS号:83-43-2)、倍氯米松(CAS号:4419-39-0)、氢化可的松(CAS号:50-23-7)、可的松(CAS号:53-06-5)、乙酰唑胺(CAS号:59-66-5)、坎利酮(CAS号:976-71-6)、氯噻酮(CAS号:77-36-1)、呋塞米(CAS号:54-31-9)、螺内酯(CAS号:52-01-7)、苄氟噻嗪(CAS号:73-48-3)、氯噻嗪(CAS号:58-94-6)、氢氯噻嗪(CAS号:58-93-5)、氨苯蝶啶(CAS号:396-01-0)、精磺胺(CAS号:121-30-2)、托拉塞米(CAS号:56211-4-6),纯度均大于等于95%,均为First Standard标准品,购于天津阿尔塔科技有限公司。
1.2 仪器与设备
8050超高效液相色谱-串联质谱(ultra-high performance liquid chromatography-tandem mass spectrometry,UPLC-MS/MS)仪 日本Shimadzu公司;3K-15型离心机 美国Sigma公司;PT2100型均质器瑞士Kinematica公司;N-EVAP112氮吹仪 美国Organomation公司;涡旋混合器 美国Scientific Industries公司;Milli-Q纯化系统 美国Millipore公司;ACQUITY UPLC BEH C18色谱柱(2.1 mmh100 mm,1.7 µm)、Oasis PRiME HLB固相萃取柱(200 mg,6 mL) 美国Waters公司;C18、中性氧化铝分散固相粉末 上海安谱实验科技股份有限公司。
1.3 方法
1.3.1 标准溶液的配制
准确称取适量各化合物标准品,用甲醇溶解,配制质量浓度约为100 μg/mL的标准储备液,于-18 ℃保存,有效期6 个月。移取各化合物标准储备液1 mL,用甲醇溶解,配制质量浓度为1.0 μg/mL的混合标准中间液,于4 ℃避光保存,有效期2 个星期。在实验过程中根据需要精确吸取适量标准溶液用甲醇-水(1∶1,V/V)稀释成所需质量浓度。
1.3.2 样品前处理
准确称取5 g样品(精确到0.01 g)于50 mL塑料离心管中,加入0.2 mol/L乙酸钠溶液(pH 5.2)8 mL,再加入β-盐酸葡萄糖醛苷酶/芳基硫酸酯酶溶液50 µL,涡旋混匀,于37 ℃下恒温水浴16 h。样品酶解后冷却至室温加入20 mL乙腈、5 g氯化钠,均质1 min,10 000 r/min离心5 min。移取10 mL乙腈层至另一离心管中,加入10 mL乙腈饱和正己烷,涡旋振荡1 min后弃去正己烷层,再加入10 mL乙腈饱和正己烷重复操作1 次。取上清液约6 mL置于50 mg C18和300 mg中性氧化铝的另一离心管中,涡旋振荡2 min,10 000 r/min离心5 min。准确移取5 mL上清液于10 mL离心管中,40 ℃氮吹至干,用1 mL体积分数50%甲醇溶液溶解残渣。经0.22 μm滤膜过滤,用UPLCMS/MS仪测定。
1.3.3 色谱条件
Waters ACQUITY UPLC BEH C18色谱柱(2.1 mmh100 mm,1.7 µm);流动相:0.005%甲酸溶液(A)和甲醇(B);流速0.4 mL/min;柱温35 ℃;进样量5 µL;梯度洗脱程序:0~1.0 min,95% A、5% B;1.0~8.0 min,95%~10% A、5%~90% B;8.0~9.0 min,10% A、90% B;9.0~9.5 min,10%~5% A、90%~95% B;9.5~11.0 min,5% A、95% B;11.0~11.1 min,5%~95% A、95%~5% B;11.1~13.0 min,95% A、5% B。
地塞米松或倍他米松检测条件:流动相:水(A)和乙腈(B);流速0.4 mL/min;柱温35 ℃;进样量5 µL;梯度洗脱程序:0~0.5 min,80%~70% A、20%~30% B;0.5~4.5 min,70% A、30% B;4.5~6.0 min,70%~10% A、30%~90% B;6.0~8.0 min,10% A、90% B;8.0~8.1 min,10%~80% A、90%~20% B;8.1~10.0 min,80% A、20% B。
1.3.4 质谱条件
电喷雾电离源(electro-spray ionization,ESI),正、负离子模式,采用多反应监测模式正负同时扫描。离子源温度350 ℃,毛细管温度250 ℃,加热模块温度350 ℃,氮气流速3.0 L/min,干燥气流速10.0 L/min,加热气流速10.0 L/min。
1.4 数据分析
所有数据均用Labsolution 5.86软件(日本Shimadzu公司)采集,采用Postrun Analysis软件进行定性分析,采用Quant Browser软件进行定量分析;采用WPS Office软件对实验数据进行柱状图绘制。
2 结果与分析
2.1 提取溶剂的选择
样品制备是药物多残留分析中的一个关键步骤,旨在同时从基质中提取出所有具有不同理化性质的分析物,有效消除基质干扰,提高方法的灵敏度和稳健性。β-受体激动剂类药物极性范围在极性至中等极性之间,以极性化合物为主的弱碱性化合物;蛋白同化激素在弱极性或中等极性的溶剂中有良好的溶解性;糖皮质激素疏水性较强;利尿剂大多同时含有酸性和碱性基团,性质差异较大。本实验在参考文献基础上,通过待测物加标回收率考察乙腈、乙酸乙酯的提取效果。乙酸乙酯回收率为42%~121%,乙腈回收率为75%~108%。相比,乙腈作为提取溶剂较为合适。
2.2 净化条件的优化
对PRiME HLB固相萃取柱和QuEChERS两种净化方式进行比较。PRiME HLB固相萃取柱不需要活化、平衡等步骤,可将提取液直接过柱达到净化目的。本实验发现,采用该种净化方式对司坦唑醇、丙酸睾酮、苯丙酸诺龙、丙酸诺龙保留性强,回收率只有20%~55%,其余化合物回收率为70.2%~116.5%。再经后续乙腈洗脱,这4 种化合物的回收率可提高至50%~80%,但洗脱液颜色加深,分析原因为部分蛋白同化剂类化合物极性较弱,易同磷脂、蛋白等一起保留在净化柱上,加大洗脱体积,连同杂质目标物洗脱出固相萃取柱,导致净化效果较差。QuEChERS前处理方法具有快速、简单、廉价、有效、可靠、安全等特点,常用的净化剂有N-丙基乙二胺、十八烷基硅烷(C18)、石墨化炭黑、中性氧化铝等。动物源食品多含有蛋白、磷脂的基质特点,实验考察不同配比C18和中性氧化铝的净化效果。取5 mL提取液,固定加入100 mg中性氧化铝,分别比较C18添加量为10、30、50、70 mg的回收率,发现C18添加量50 mg时46 种化合物的回收率最高;固定加入50 mg C18,分别比较中性氧化铝添加量为100、300、500 mg的回收率,发现中性氧化铝添加量300 mg时46 种化合物的回收率最高。因此,实验最终选择提取液加入50 mg C18和300 mg中性氧化铝作为组合净化剂,可以获得良好的净化效果,基质干扰小,且不会对目标物产生明显吸附。
2.3 色谱条件的优化
本实验所测化合物性质差异较大,且含有3 对同分异构体,坎利酮和螺内酯、地塞米松和倍他米松、泼尼松龙和可的松。这3 对化合物具有相同的前体和产物离子,若要准确定量这些化合物,色谱峰需有一定程度的分离。因此,实验对色谱参数如流动相梯度条件、流动相成分进行测试,以实现目标化合物的最佳色谱分离以及各化合物的良好峰形和最佳响应。实验考察首先考察甲醇、乙腈、0.1%甲酸溶液、水作为流动相,采用不同梯度在ACQUITY BEH C18色谱柱(100 mmh2.1 mm,1.7 µm)上进行分离,结果显示,无论在乙腈-水还是甲醇-水条件下,泼尼松龙和可的松均可达到分离,但采用甲醇不能分离地塞米松和倍他米松,在乙腈条件下坎利酮和螺内酯分离度也很差,需要运行36 min才能彻底分离,并且峰形较宽。综合其他化合物,在乙腈-水条件下西马特罗、特布他林、齐帕特罗峰溶剂效应较强,峰提前流出,司坦唑醇峰形较差。在甲醇-水条件下,除司坦唑醇峰形较差外,各化合物在色谱柱上保留、峰形均较好。与乙腈-水体系相比,甲醇-水条件下喷布特罗、丙酸睾酮和苯丙酸诺龙响应值提高2~5 倍。加入0.1%甲酸,β-激动剂、坎利酮、螺内酯、睾酮等化合物灵敏度提高0.5~2 倍,苄氟噻嗪、氯噻嗪、乙酰唑胺和糖皮质激素类等大部分化合物电离产生抑制作用,但司坦唑醇峰形得到改善。通过再次优化甲酸浓度,实验最终采用0.005%甲酸-甲醇作为流动相,除地塞米松和倍他米松无法分离外,各化合物灵敏度和和峰形均可满足要求,整个梯度运行时间13 min;当地塞米松和倍他米松检出时采用1.3.3节色谱条件分离进行准确定量。46 种化合物的总离子流色谱图见图1。

图1 46 种食源性兴奋剂标准溶液总离子流色谱图(10 ng/mL)Fig.1 Total ion current chromatograms of 46 foodborne stimulant drugs (10 ng/mL)
2.4 质谱条件的优化
采用不接分析柱的方式向质谱系统注入1 mg/L标准溶液,进样量1 μL。确定各化合物的最佳质谱条件,包括离子源温度、毛细管温度、加热模块温度、加热气流速、干燥气流速、氮气流速、选择特征离子对、Q1和Q3电压、碰撞能量等质谱分析条件。各化合物标准溶液和UPLC流动相溶液混合进入ESI源,在正负离子同时扫描方式下进行一级全扫描质谱分析,其中大部分化合物分子离子峰[M+H]+响应值最高,噻嗪类利尿剂均表现为[M-H]-响应值最高。螺内酯在高温离子源内不稳定,脱去一分子乙酰基硫基可显示较高的[M-C2H3OS]+,与坎利酮母离子相同。可的松和泼尼松龙的分子离子峰[M+COOH]-峰反而比[M+H]+响应值高。然后选择相应的母离子峰进行子离子扫描,得到主要的子离子,并选择响应值高的子离子进行碰撞能量优化,最终确定多反应监测的离子对及碰撞能量,本实验采用正、负离子切换模式同时检测样品中46 种兴奋剂类药物。建立的质谱参数如表1所示。

表1 46 种化合物的质谱参数、线性关系、检出限和定量限Table 1 Mass spectrometric parameters, linearity, LODs and LOQs of 46 compounds

续表1

续表1
2.5 基质效应
基质效应是指色谱分离时共洗脱的物质改变了待测成分的离子化效应,引起信号的抑制或提高。基质效应影响大时会降低方法的灵敏度和准确性,造成测定误差。动物源性食品基质复杂,容易对目标物产生干扰。一般认为,基质匹配校正曲线斜率与标准曲线斜率的比值在0.85~1.15之间不存在基质效应[15-17]。本实验方法对所选动物源样品存在一定基质效应,以猪肉为例,基质效应在0.32~1.40之间,结果见图2。所建立方法采用基质标准溶液进行校正。

图2 46 种食源性兴奋剂基质效应Fig.2 Matrix effects of 46 foodborne stimulant drugs
2.6 方法学验证
2.6.1 方法的线性关系、检出限和定量限
取不含46 种化合物的猪肉、牛肉、羊肉、鸡蛋和牛奶按照1.3.1节方法制备得到空白基质溶液,用各基质溶液稀释混合标准中间液得到质量浓度为0.1、0.5、1.0、2.0、5.0、10、20、50、100、200 ng/mL系列混合标准溶液,绘制基质标准工作曲线,得到各化合物线性范围、线性方程和相关系数(r)。以分析物质量浓度为横坐标,相应峰面积为纵坐标,结果显示,46 种化合物的基质标准工作液在各自范围内均呈良好线性关系,相关系数均大于0.99。
采用信噪比(RSN)法进行46 种目标化合物的检出限和定量限确定,在各基质空白样品中添加标准溶液按1.3.2节步骤进行样品前处理后进样测定,以RSN≥3计算检出限,RSN≥10计算定量限,同一化合物在不同基质中检出限、定量限和线性范围一致(表1)。代表性牛肉基质中各化合物的线性方程和相关系数见表1。
2.6.2 方法的回收率和精密度
根据GB/T 27404ü2008《实验室质量控制规范 食品理化检测》的要求,实验选取猪肉、牛肉、羊肉、鸡蛋和牛奶5 种不含46 种食源性兴奋剂化合物的基质分别在3 个质量浓度进行加标回收实验,每个水平测定6 次。由表2可知,方法的平均回收率在76.0%~111.9%之间,相对标准偏差(relative standard deviation,RSD)在2.3%~12.4%之间。

表2 方法的加标回收率及RSD(n=6)Table 2 Recoveries and RSDs of the developed method (n = 6)

续表2
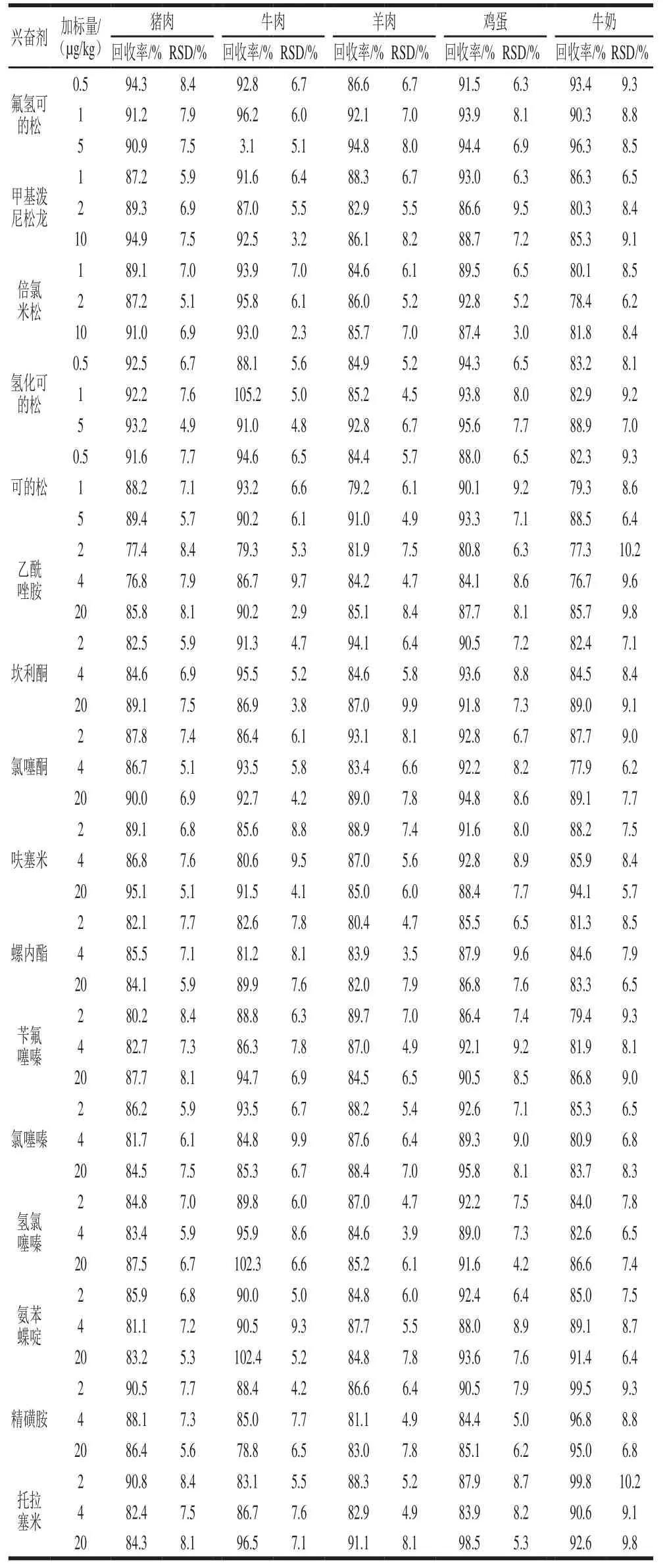
续表2
2.7 实际样品分析
采用本方法对市售猪肉、牛肉、羊肉共计50 批,鸡蛋10 批和牛奶10 批进行测定,结果表明,1 批猪肉检出克伦特罗,检出量为1.86 µg/kg,采用GB/T 22286ü2008《动物源性食品中多种β-受体激动剂残留量的测定 液相色谱串联质谱法》[23]进行验证,结果为1.90 µg/kg,RSD为1.50%,无明显差异,结果可靠。克伦特罗阳性样品离子色谱图和质谱图如图3所示。

图3 阳性样品中克伦特罗提取离子色谱(A)及质谱图(B)Fig.3 Extracted ion chromatogram (A) and mass spectrum (B) of clenbuterol in positive samples
2.8 与文献方法比较
如表3所示,本方法采用普适性较强的UPLC-MS/MS仪建立动物源性食品中10 种β-受体激动剂、3 种β-受体阻断剂、13 种蛋白同化剂、9 种糖皮质激素、11 种利尿剂共计5 类46 种食源性兴奋剂的同时检测技术,定量限为0.1~2.0 μg/kg,可满足大型体育赛事对于食源性兴奋剂的监管要求。与文献方法相比,该方法普适性强,覆盖化合物范围全面,测定基质范围广泛,可以一次样品处理获得所有结果,提高工作效率。

表3 本方法与文献分析方法的对比Table 3 Comparison of the present method with previously reported methods
3 结 论
应用QuEChERS净化,以UPLC-MS/MS法建立同时检测动物源性食品中46 种食源性兴奋剂的方法,分别对前处理方法、色谱和质谱条件进行优化,采用多反应监测模式检测,测定β-激动剂、糖皮质激素类、利尿剂类和蛋白同化剂类等多种食源性兴奋剂。该方法具有灵敏度高、普适性强、操作简便等优点,适用于动物源性食品中多种食源性兴奋剂的测定,可为体育赛事的食品保障工作提供有效的技术支撑。
猜你喜欢
中老年保健(2022年1期)2022-08-17
煤化工(2022年3期)2022-07-08
体育科技文献通报(2022年3期)2022-05-23
合成纤维工业(2021年5期)2021-10-31
食品安全导刊(2021年21期)2021-08-30
食品安全导刊(2021年20期)2021-08-30
中老年保健(2021年6期)2021-08-24
成都体育学院学报(2021年1期)2021-07-16
首都食品与医药(2020年1期)2020-10-21
山东工业技术(2016年10期)2016-09-06
