
基于CRISPR/Cas9系统构建Slfn2基因敲除的小鼠肺癌细胞系
2022-08-08 05:37周祖平蒲仕明
广西师范大学学报(自然科学版) 2022年4期
陈 莹,周祖平,3,邢 兵,蒲仕明,3*
(1.广西师范大学 生命科学学院, 广西 桂林 541006;2.广西高校干细胞与医药生物技术重点实验室(广西师范大学),广西 桂林 541004;3.广西师范大学 生物医学研究中心,广西 桂林 541004)
肺癌(lung cancer)是全球范围内发病率、死亡率最高的恶性肿瘤,其中近一半病例(49.9%)发生在发展中国家[1]。肺癌是全球男性最常见癌症且是男性癌症死亡的主要原因[2]。我国肺癌发病率和死亡率位居所有癌症之首,每年新发肺癌病例约78.7万例,每年因肺癌死亡例数为63.1万[3]。
Schlafen(SLFN)蛋白家族最初在小鼠细胞中被发现,该家族蛋白在一些恶性肿瘤中可以抑制癌细胞的迁移和入侵[4-6]、肿瘤锚定依赖生长[7-9]、促进癌细胞对化疗的敏感性[6,11-12],表明SLFNs家族蛋白在肿瘤领域有可能成为一种新的治疗靶点。SLFN2作为SLFNs的关键成员,调节T细胞的活化[12-13]。刘梦伊等[14]研究表明肿瘤细胞中Slfn2基因稳定表达后,其生长增殖速度明显减缓,迁移能力也显著降低,提示Slfn2基因在抑制肿瘤发生过程中发挥重要作用。目前Slfn2基因参与肿瘤细胞活动的作用及分子机制尚不清楚,为深入研究Slfn2基因在肿瘤发生发展中的重要作用,本文利用CRISPR/Cas9技术构建Slfn2-/-LLC细胞,为开展后续机制研究的相关实验奠定基础。
1 材料与方法
1.1 材料
1.1.1 生物材料
肺癌细胞系LLC、胚肾细胞293T购自中科院典型培养物昆明细胞库;感受态Stb13、pLenti CRISPR v2、pMD2.G、psPAX2购自武汉淼灵生物科技有限公司;质粒测序由天一辉远生物有限公司完成。
1.1.2 主要试剂
T4连接酶、Esp3I(BsmBI)酶、胎牛血清、DMEM、Agarose、单链DNA oligo与引物合成均购自Thermo Fisher Scientific;无内毒素质粒小提中量试剂盒、氨苄青霉素、嘌呤霉素、PCR mix购自天根生化科技有限公司。
1.1.3 主要仪器
PCR仪器,购自Biometra公司;恒温水槽,购于上海力辰邦西仪器科技有限公司;恒温摇床,购于上海驸玛实验设备有限公司;CO2恒温培养箱,购于Thermo Fisher公司;霉菌培养箱,购于上海智城分析仪器制造有限公司;水平电泳槽、通用电泳仪、凝胶成像系统均购于Bio-Rad公司。
1.2 方法
1.2.1 sgRNA序列的设计
在CHOPCHOP基因编辑网站(http:∥chopchop.cbu.uib.no/)的“Mus muscμLus(mm10/GRCm38)”物种下查询Slfn2(NM_011408.1)基因“CRISPR/Cas9”的全部“Knock-down”突变位点,选取脱靶预测最低的2个sgRNA序列,并根据pLenti CRISPR v2酶切位点黏性末端序列设计oligo与之对应的黏性末端,即在正义链5′端添加CACC,反义链5′端添加AAAC(图1)。此外,sgRNA的5′端不为“G”的则额外添加“G”。
1.2.2 pLenti CRISPR-Slfn2组质粒的构建
1)oligo的退火。将合成的oligo sgRNA稀释至50 μmol/L,各取10 μL与Annealing Buffer(5×,10 μL)混合并定容至50 μL;随后加热至95 ℃后缓慢降温至25 ℃。稀释200倍后使用(50 nmol/L)。
2)sgRNA表达。载体酶切重组的酶切连接反应体系:pLenti CRISPR v2(100 ng/μL)1 μL、oligo(50 nmol/L)1 μL、T4 DNA Ligase 1 μL、5× DNA Ligase Reaction Buffer 4 μL、Esp3I 1 μL、10× Buffer Tango 2 μL、DTT(20 mmol/L)1 μL、ddH2O 9 μL,总体积20 μL。将20 μL反应物置于PCR仪,按程序进行酶切连接反应:37 ℃ 3 min、23 ℃ 3 min, 20个循环;37 ℃ 10 min,50 ℃ 30 s,80 ℃ 5 min,4 ℃ 无限循环。
3)重组质粒转化。将构建的质粒4 μL加入至50 μL Stb13感受态细胞中,轻微混匀后冰上孵育30 min,随后将感受态置42 ℃水浴热激90 s,并快速将其转移到冰上冰浴5 min。向每管感受态中加入500 μL LB培养基后,将其转移到37 ℃摇床培养箱(200 r/min)恒温培养1 h。
4)重组质粒鉴定。将感受态10 μL均匀涂抹到含氨苄青霉素的固体LB培养基上,并将培养皿转移到37 ℃霉菌培养箱培养12~16 h。挑取良好的菌落加入到PCR体系(含PCR mix、Primer-pLenti-F:GGTACCGAGGGCCTATTTCC,Primer-pLenti-R:ACTTTCCCAGTTTACCCCGC)中,对插入sgRNA区域的片段扩增,获取扩增片段进行电泳验证和测序验证。
1.2.3 质粒提取
将验证的菌落活化后,加入到含氨苄青霉素的液体LB培养基中,置37 ℃摇床培养箱(300 r/min)培养12~16 h。质粒提取严格按照天根无内毒素质粒小提中量试剂盒(DP118-02)实验流程进行。提取的质粒测定浓度后待用。pMD2.G、psPAX2质粒的转化、扩大、提取均按1.2.2节方法进行。
1.2.4 慢病毒包装
首先,将pLenti CRISPR v2、pMD2.G、psPAX2按摩尔比1∶1∶1(共30 μg)加入到DMEM培养基中(最终体积为500 μL),充分混匀制备成质粒体系;将90 μL PEI(1 g/L)加入到410 μL DMEM培养基中,充分混匀制备成转染体系。接着,两体系室温孵育5 min后,将质粒体系逐滴加入涡旋震荡的转染体系中,并室温孵育20 min。随后,经转染体系逐滴加入生长良好且细胞融合度为70%~80%的293T细胞中转染4 h。然后,去除转染体系,加入DMEM完全培养基(含体积分数为10%FBS、体积分数为1%双抗,下同)在二氧化碳培养箱(37 ℃、体积分数5%CO2,下同)培养12 h后,换新鲜的DMEM完全培养基继续培养48 h。最后,收取病毒液,并用0.45 μm过滤器过滤病毒液。病毒液经慢病毒滴度快速检测卡(购于北京博奥龙免疫技术有限公司)检测病毒滴度后待用。
1.2.5 慢病毒转染LLC细胞
将病毒液加入到生长良好、细胞融合度为30%的LLC细胞中感染48 h。随后将培养基换成含有嘌呤霉素(终浓度为4 mg/L)的DMEM完全培养基,筛选2个48 h。
1.2.6 单克隆培养
将筛选得到的LLC细胞经消化后,调整细胞浓度为1×105个/mL,用有限稀释法将细胞接种到96孔板中。待细胞贴壁后用显微镜观察含有1个细胞的培养孔,并做好标记。
1.2.7 细胞DNA提取
单克隆细胞扩大后,取部分细胞加入细胞裂解液(10 mmol/L Tris-Cl、0.1 mol/L EDTA、0.5% SDS、10 mg/L蛋白酶k)裂解2 h,加入2倍体积乙醇混匀,15 000×g离心沉淀DNA,经体积分数70%乙醇清洗2次后,加入适量ddH2O溶解DNA。
1.2.8 目的基因敲除鉴定
将提取的DNA加入到PCR体系(含PCR mix、Primer-Slfn2-F:CTGGGAAAATGGGCATCAGTG,Primer-Slfn2-R:CAGCTCCGGATTTGTGCACTT)中,对敲除区域的片段进行扩增,获取扩增片段进行电泳验证和测序验证。
2 结果
2.1 sgRNA序列设计
在CHOPCOHP网站选取脱靶率低、编辑效率较高且位于Slfn2外显子上的sgRNA位点2个(sgRNA-1:GCTCTTTGATGCGTTTTCGC,sgRNA-2:TGTGCTGTCCTGAATTCGGG),并结合酶切位点设计和合成oligo序列(图1)。
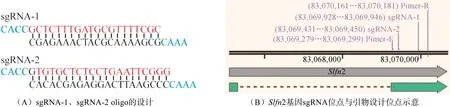
蓝色部分为插入接头序列,红色部分为sgRNA,黑色部分为sgRNA的反向互补序列
2.2 pLenti CRISPR-Slfn2重组质粒的构建
针对Slfn2基因序列,设计2个sgRNA寡核苷酸序列,将sgRNA与pLenti CRISPR v2连接后,转化至Stb13感受态细胞中,经筛选后对PCR产物进行验证。结果表明,pLenti CRISPR v2原始质粒大小为2 374 bp,而经过构建的质粒pLenti CRISPR-Slfn2-1&2的PCR产物大小为514 bp(图2(A)、(B))。随后对pLenti CRISPR-Slfn2-1&2 PCR产物测序,结果表明,sgRNA序列已经成功结合到pLenti CRISPR v2中,pLenti CRISPR-Slfn2-1&2重组质粒构建成功(图2(C))。
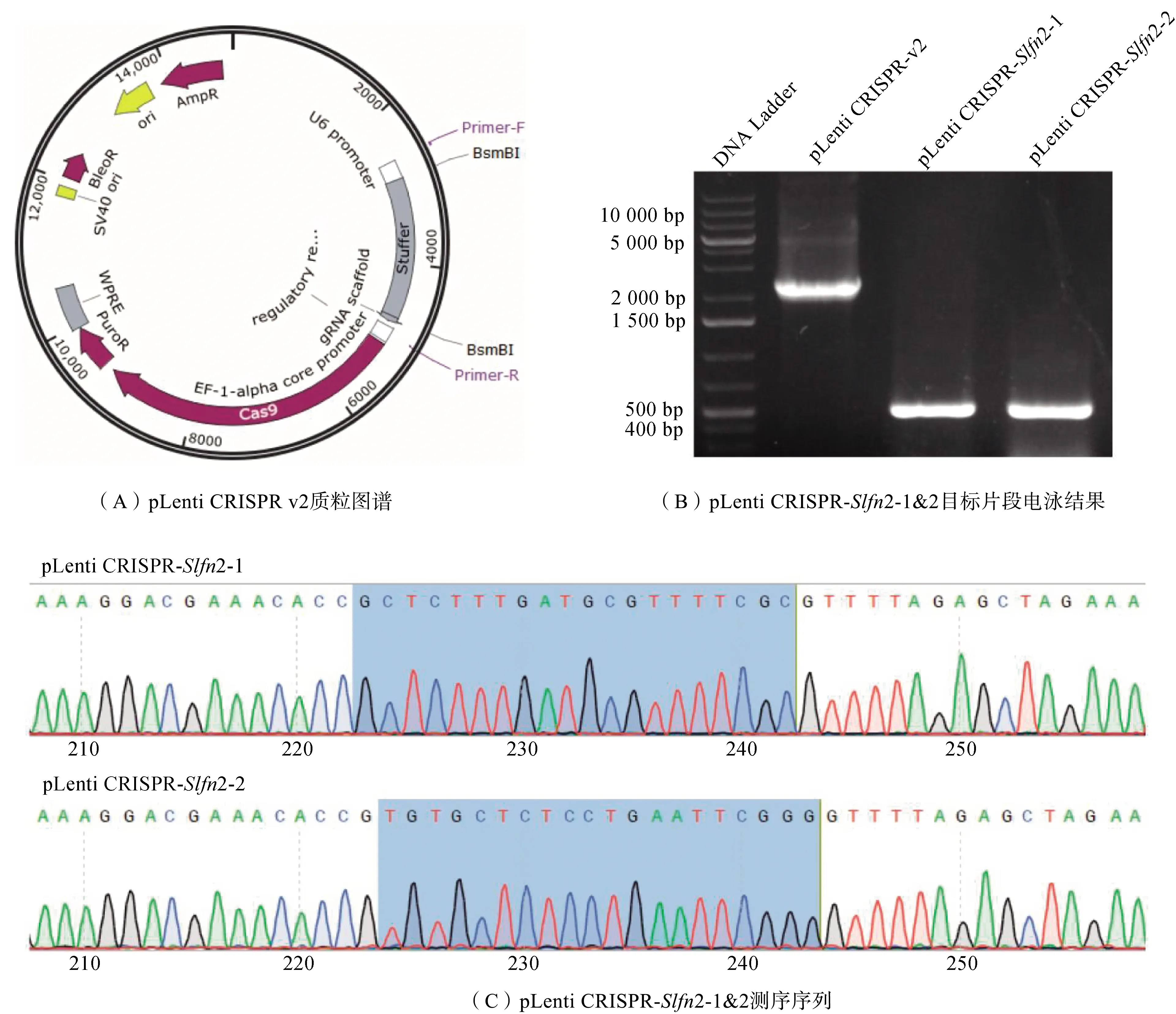
图2 pLenti CRISPR-Slfn2重组质粒的验证
2.3 突变克隆筛选与鉴定
将重组质粒、pMD2.G、psPAX2转染进入293T细胞包装病毒,利用收集的病毒感染LLC细胞。经筛选后,进行单克隆培养,并对4个单克隆(Slfn2-1a、Slfn2-1b为sgRNA-1位点突变的单克隆,Slfn2-2a、Slfn2-2b为sgRNA-2位点突变的单克隆)进行PCR和测序验证。结果表明:Slfn2-ctr扩增的PCR片段大小为691 bp,Slfn2-1a、Slfn2-1b、Slfn2-2a扩增的PCR片段均无明显碱基丢失,Slfn2-2b的PCR片段有明显的碱基丢失(图3(A))。Slfn2-2a测序结果表明,在sgRNA-2剪切处出现了8个碱基丢失,造成移码突变(图3(B)、(C))。Slfn2-2b测序结果表明,在sgRNA-2剪切处出现了180个碱基丢失,造成60个氨基酸的丢失突变(图3(D)、(E))。Slfn2-1a、Slfn2-1b在sgRNA-1突变位点处出现碱基测序混乱,可能获得非纯净的单个克隆。这些结果证明利用sgRNA-2获得了2个Slfn2基因敲除LLC细胞系。
3 讨论
Liu等[6]研究发现,肿瘤细胞中Slfn2基因稳定表达后,其生长增殖速度明显减缓,迁移能力也显著降低,提示Slfn2基因在抑制肿瘤发生过程中发挥重要作用。为深入研究Slfn2在肿瘤细胞中的作用及分子机制,本文利用pLenti CRISPR v2构建的重组质粒与pMD2.G、psPAX2包装病毒,并利用病毒实现LLC细胞中Slfn2的敲除,构建Slfn2-/-LLC细胞用于后续研究。
Slfn2共5 567 bp,含有2个外显子,共379个氨基酸,其中第2个外显子含362个氨基酸,突变位点的选择位于第2个外显子上。经筛选后,获得sgRNA-1、sgRNA-2这2个不同突变位点的SgRNA,其中CHOPCHOP网站预测突变效率sgRNA-1为48.11%,sgRNA-2为67.27%。在实验获取的单克隆中,sgRNA-1的单克隆并没有获得单一的细胞突变,测序后突变位点处序列混乱,可能是制备单克隆后实现的部分细胞突变;而突变效率较高的sgRNA-2的单克隆获得单一的细胞突变。sgRNA-2获得的2个单克隆中,Slfn2-2a剪切处出现了8个碱基丢失。8个碱基的缺失不仅仅丢失了2个氨基酸,更重要的是实现突变位点后外显子的移码突变(图3(B)、(C))。Slfn2-2b在剪切处出现了180个碱基丢失,造成60个氨基酸的缺失突变(图3(D)、(E))。此外,sgRNA-1位于第2个外显子的下游,形成的移码突变区域小,而sgRNA-2位于第2个外显子的上游,形成的移码突变Slfn2-2a剪区域较大。
4 结语
本研究应用CRISPR/Cas9基因编辑技术,实现Slfn2基因在LLC细胞中的敲除,并获得移码突变和60个氨基酸序列缺失的Slfn2-/-LLC细胞各1株,为后续深入研究Slfn2在肿瘤细胞中的作用及分子机制提供稳定的研究对象。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
中国生殖健康(2020年4期)2021-01-18
中国现代医药杂志(2020年10期)2020-12-14
教学考试(高考生物)(2020年6期)2020-11-23
食品与生物技术学报(2020年8期)2020-01-06
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
中国生殖健康(2018年4期)2018-11-06
现代检验医学杂志(2016年3期)2016-11-15
医学研究杂志(2015年3期)2015-06-10
