
南海近岸硬骨鱼鳃组织的细菌群落及多样性分析
2022-08-23 07:36郭迎香杨李玲许友伟方艺菲姜敬哲
南方水产科学 2022年4期
郭迎香,杨李玲,许友伟,方艺菲,3,王 萌,姜敬哲
1. 天津农学院 水产学院,天津 300384
2. 中国水产科学研究院南海水产研究所/农业农村部南海渔业资源开发利用重点实验室,广东 广州 510300
3. 上海美吉生物医药科技有限公司,上海 201318
4. 广州市从化区农业农村局,广东 广州 510925
南海又称南中国海或中国南海,是世界第三大边缘海[1],拥有丰富的海洋生物资源。同时,南海也是海上“丝绸之路”的起点和国家海洋强国战略的“主战场”,具有重要的生态、经济和政治意义。但20世纪80年代以来,南海渔业资源受海洋污染和过度捕捞的双重影响而不断衰退[2]。因此自1999年起,我国政府开始对南海区域渔船数量等进行管控,调整渔业生产结构,以降低南海渔业资源的开发强度[3],保护海洋渔业资源。
鱼类是海洋中最主要的动物类群,现存种类主要包括硬骨鱼纲、软骨鱼纲、盲鳗纲和头甲纲4个纲[4]。张俊等[5]发现南海中南部中层鱼中硬骨鱼纲的种类最多,是南海海洋生态系统的主要物种。鳃是鱼类等海洋生物的主要呼吸器官,除了具有呼吸作用,还具有滤食、渗透压调节、pH平衡调节和氨排泄等功能[6]。同时,鳃也是海洋环境微生物进入宿主体内的第一道关卡,对宿主内生的微生物群落具有重要调控作用。海洋鱼类鳃组织中的微生物类群是一个重要的免疫屏障,也是鱼类抵御海洋环境中潜在病原体的第一道防线。研究发现,鱼类鳃组织中的细菌与宿主的疾病具有重要的相关性,Toenshoff等[7]通过对大西洋鲑 (Salmon salar) 鳃组织菌群研究发现了一种新型的β-proteobacterial会引起宿主鱼体上皮囊炎等疾病。因此,对鳃组织上附着的微生物群落进行研究,对更好地理解海洋动物在海洋环境中的适应性作用、以及病原入侵与鱼类病害的发生机制具有重要意义。近年来,海洋鱼类微生物研究大多关注在各种鱼类的肠道组织,如日本花鲈 (Lateolabrax japonicus)[8]、大西洋鳕(Gadus morhua)[9]、大西洋鲑[10]、大菱鲆 (Scophthalmus maximus)[11]和大黄鱼 (Larimichthys crocea)[12]等经济性物种,而围绕鳃组织微生物多样性及其功能的研究匮乏。王小彤和许强华[13]报道了4种南极鱼类鳃组织微生物群落多样性,Pratte等[14]对珊瑚礁不同鱼类鳃组织微生物群落多样性进行了报道。但相较于种类繁多的海洋鱼类资源,鳃组织微生物的研究数据还十分匮乏。
16S rDNA (或 16S rRNA) 基因作为细菌的保守特征序列是进行细菌系统发育和分类鉴定的标签基因[15]。随着高通量测序成本的降低和扩增子 (Amplicon) 技术的日臻完善,16S rDNA扩增子已逐渐成为微生物多样性研究的主流方法[16]。本研究基于16S扩增子技术,对从南海近岸海域7个站点采集的45种海洋硬骨鱼纲生物的鳃组织样品进行了细菌群落分析,以期为了解影响海洋动物鳃组织微生物群落的主要因素、探索海洋动物与海洋环境微生物的相互依存关系提供参考。
1 材料与方法
1.1 样品采集与分组
2019年3月,在7个站点共采集了52份鱼类鳃组织样品 (分类见表1),分属于11目33科40属的45种硬骨鱼纲鱼类。7个站点的位置见图1,其中D3、D8、F3、F8、H3、H8和I9站点距离海岸的距离分别为60、215、10、175、20、130和165 km。采集到的鲜活样品迅速装于无菌密封袋中,于−80 ℃暂存。上岸后用无菌解剖工具完整取下鳃组织并于液氮中速冻后,置于−80 ℃长期保存。本实验将同一站点采集的同种鱼类的鳃组织,根据鱼体大小,进行1~3尾的混合而作为一个样品,以保证文库构建和测序数据的质量。

表1 样品分类信息Table 1 Taxonomy information of samples
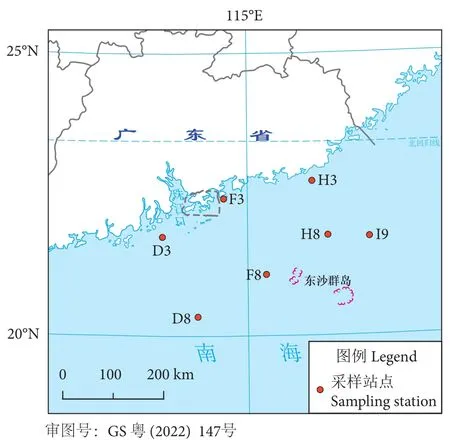
图1 广东近岸南海海域采样站点示意图Fig. 1 Diagram of sampling stations in Guangdong coastal waters in South China Sea
分别按照采样站位、宿主来源可将52个样品进行两种分组编排。依站点不同可分为7组,依宿主目水平可分为3组,其中鲈形目样品最多 (19个),鲉形目16个,其他小目统一编为一组 (17个)。
1.2 细菌 DNA 提取、文库构建和测序
使用 HiPure Bacterial DNA kit (Magen,广州)试剂盒,参照说明书进行总DNA提取,利用Thermo NanoDrop One (Thermo Fisher Scientific Inc., 上海) 检测DNA的纯度和浓度。针对16S rDNA基因的V4区进行巢套式扩增,使用带barcode 的特异引物及 TaKaRaPremix Taq® Version 2.0(TaKaRa Biotechnology Co., 大连) 进行 PCR 扩增。所用一轮引物为27F (5'-AGAGTTTGATCCTGGCT CAG-3') 和 1492R (5'-TACGGYTACCTTGTTAY GACTT-3'),二轮引物为 515F (5'-GTGCCA GCMGCCGCGGTAA-3') 和 806R (5'-GGACT ACHVGGGTWTCTAAT-3'),扩增区域长度为 310 bp。一轮 PCR 反应体系为 (20 μL):Premix TaqTM(Ex TaqTMVersion 2.0 plus dye)/Mix 10 μL,上下游引物(10 μmol·L−1) 各 0.4 μL,模板 DNA (40~60 ng) 各1 μL,Nuclease-free water 8 μL。二轮 PCR 反应体系(50 μL) 为:Premix TaqTM(Ex TaqTMVersion 2.0 plus dye)/Mix 25 μL,上下游引物 (10 μmol·L−1) 各 1 μL,模板 DNA (40~60 ng) 1 μL,Nuclease-free water 22 μL。PCR 反应条件为 94 ℃ 5 min;94 ℃ 30 s,53 ℃ 30 s,72 ℃ 30 s,30 个循环;72 ℃ 8 min。DNA 提取、文库构建及测序工作委托广东美格基因科技有限公司进行。
1.3 高通量测序及数据分析
采用 Illumina Nova 6000平台对构建的扩增子文库进行PE250测序 (广东美格基因科技有限公司,广州),测序完成后得到原始的下机数据:双端原始序列 (Paired-end Raw Reads),利用 fastp[17](An ultra-fast all-in-one FASTQ preprocessor, V 0.14.1,https://github.com/OpenGene/fastp) 分别对两端的原始序列进行滑窗 (sliding window) 质量剪裁,同时根据序列首尾两端的引物信息利用cutadapt软件(https://github.com/marcelm/cutadapt/) 去除引物,得到质控和过滤后高质量的双端优化序列 (Clean Reads)。对于双端优化序列,根据reads之间的overlap关系,利用 usearch-fastq_mergepairs (V10,http://www.drive5.com/usearch/预设参数包含最小overlap长度设置为16 bp,拼接序列的overlap区允许的最大错配5 bp等) 过滤不符合的Tags,获得原始的拼接序列 (Raw Tags),利用fastp对拼接序列数据进行滑窗质量剪裁,得到优化的拼接序列(Clean Tags)。利用UPARSE[18]方法进行 OTU 聚类。利用usearch-sintax将每个OTU的代表序列与 SILVA (16S)、RDP (16S)、Greengenes (16S) 数据库进行比对获得物种注释信息,以达到了解所有序列物种来源的目的,最终用SILVA (16S) 数据库注释得到的OTU结果进行后续分析。
1.4 细菌群落多样性
使用过滤后获得的最终OTU丰度表进一步进行物种群落组成分析、Alpha多样性分析、Beta多样性分析。Alpha多样性使用物种丰富度或均匀度来表征,其中 Chao1[19]、 ACE 和 Observed species指数用于估算群落物种丰富度[20]。而Shannon[21]和Simpson指数用于反映群落物种多样性。Beta多样性通过非度量多维尺度分析 (Nonmetric Multidimensional Scaling, NMDS) 来表征,NMDS 可以基于进化关系或数量距离矩阵对比样本之间的差异。
1.5 细菌群落功能预测
使用PICRUSt软件[22]预测OTU的功能,首先消除16S marker gene在物种基因组中的拷贝数的影响,然后通过每个OTU所对应的greengene id,分别比对到京都基因和基因组百科全书 (Kyoto Encyclopedia of Genes and Genomes, KEGG) 数据库和 COG (Clusters of Orthologous Groups) 数据库,获得功能预测信息,根据OTU丰度计算各功能类别的丰度,分别得到各样本在不同分类水平的功能丰度表。
使用在线微生物组分析平台MicrobiomeAnalyst (https://www.microbiomeanalyst.ca) 进行微生物数据的多样性和功能分析[23]。
2 结果
2.1 测序数据概述
对52份16S rDNA文库进行测序,共获得有效拼接序列 2 952 366 条,平均每个文库 56 776 条;共产生10 617个OTU。图2表明各样本稀释曲线随着测序深度的增加逐渐趋于平缓,所有样品均达到平台期,表明本研究的测序深度足够。由稀释曲线的位置可知,I9站点样品的OTU数量相对较高,D8站点样品较低。
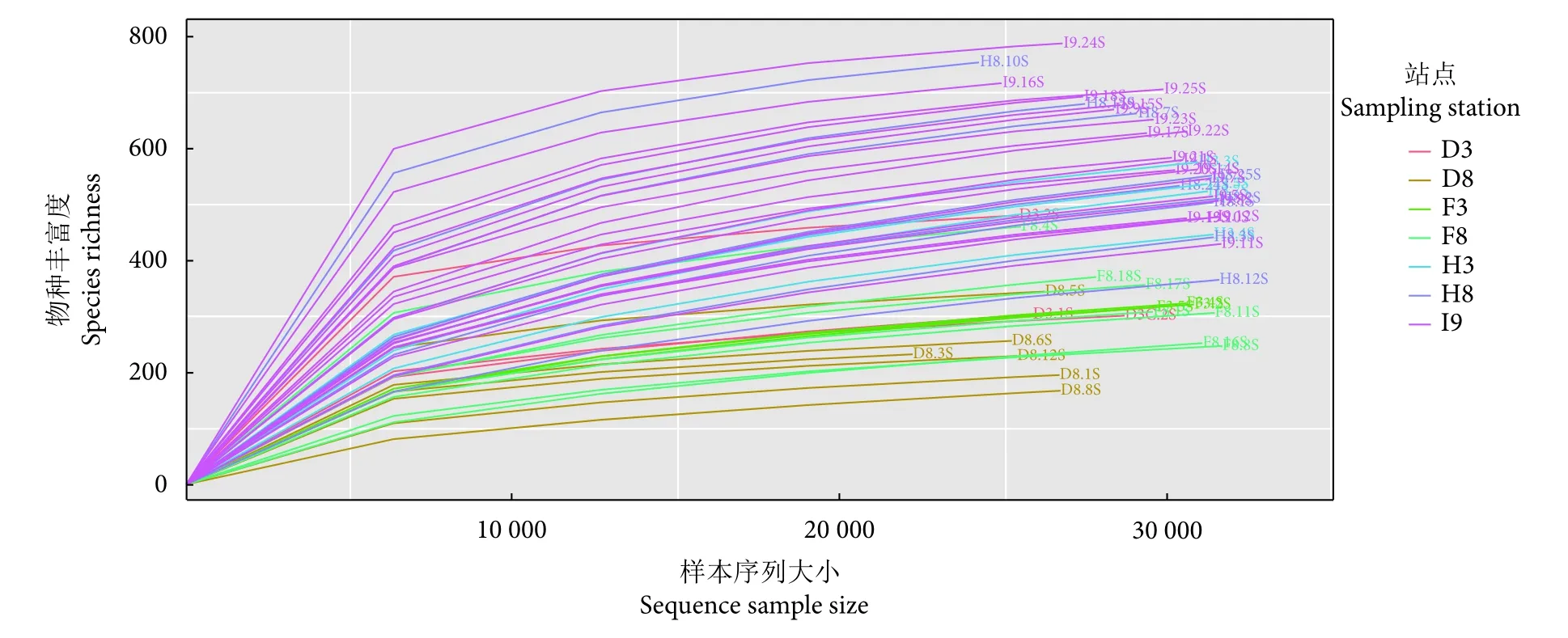
图2 各个样本的稀释曲线Fig. 2 Rarefaction curve of each sample
2.2 鱼鳃细菌群落组成
2.2.1 门水平组成
对不同站点样品的细菌门水平分类组成和相对丰度情况进行了统计 (图3)。在总体水平上,平均相对丰度≥1%的优势菌门依次为变形菌门 (Proteobacteria, 71.3%)、厚壁菌门 (Firmicutes, 10.3%)、浮霉菌门 (Planctomycetes, 8.3%)、放线菌门 (Actinobacteria, 3.4%)、拟杆菌门 (Bacteroidetes, 2.1%) 和蓝细菌 (Cyanobacteria, 1%)。不同站点的优势菌门丰度不同,但变形菌门均为所有站点中丰度最高的优势类群,其中以F3和F8站点最高,D3站点最低。厚壁菌门主要分布于D3、D8、I9站点,而浮霉菌门主要分布于D3、H8、I9站点,其他门在不同站点间丰度比例略有差异。另外,F3和F8站点的细菌种类较少,而H8和I9站点的细菌种类相对较多。
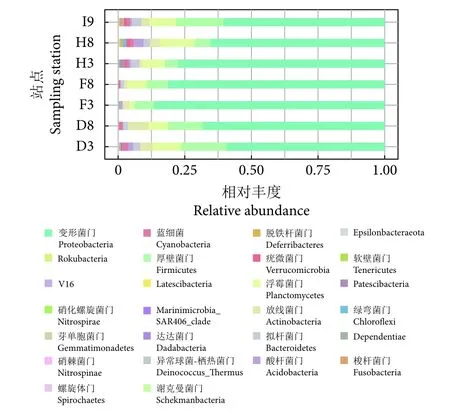
图3 鱼鳃细菌门水平物种组成相对丰度Fig. 3 Relative abundance of taxonomy composition of bacteria in fish gill at phylum level
2.2.2 属水平组成
进一步在属水平上对各站点样品的分类组成和相对丰度进行统计分析。在所有站点中,相对丰度≥2%的优势菌属依次为不动杆菌属 (Acinetobacter, 17.2%)、青枯菌属 (Ralstonia, 7.5%)、嗜冷杆菌属 (Psychrobacter, 7.5%)、鞘氨醇单胞菌 (Sphingomonas, 3.5%)、BD1_7_clade(3.3%) 和Clostridium_sensu_stricto_1 (3%,图4)。不同菌属在不同站点的丰度占比不同,但不动杆菌属在所有站点中均为优势菌属。青枯菌属 (26.5%) 和鞘氨醇单胞菌属 (20.5%) 主要分布于 F3站点,BD1_7_clade在F8站点占比最高 (20.3%),而嗜冷杆菌属在H3(20.7%) 和 I9 站点 (17.9%) 丰度较高。总体来看,D8、H8、I9站点的细菌在属水平上的种类相对其他几个站点较多,各个站点优势菌属的相对丰度相差较大。
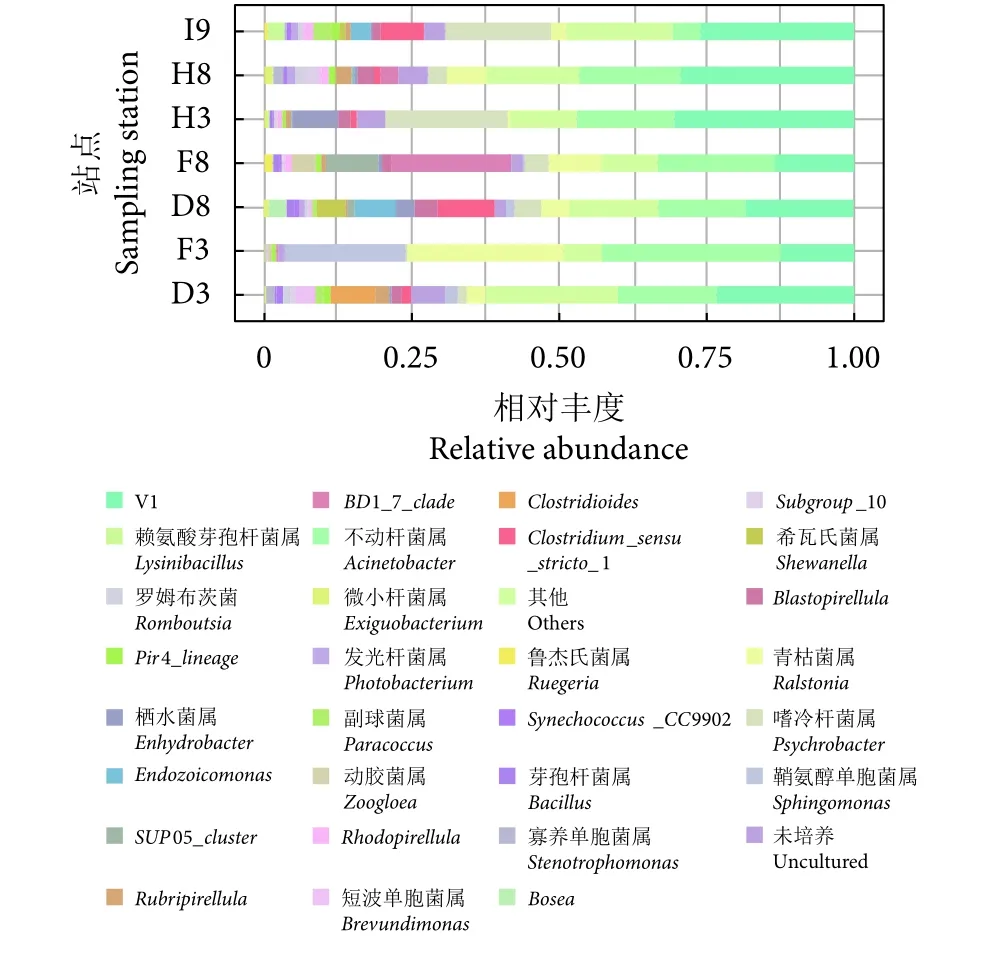
图4 鱼鳃细菌属水平物种组成相对丰度Fig. 4 Relative abundance of taxonomy composition of bacteria in fish gill at genus level
2.3 Alpha 多样性分析
Alpha多样性指数可用于反映样品内部微生物群落的相对丰度及多样性。本研究计算了各站点分组样品的Alpha多样性指数 (图5)。结果表明,H3、H8和I9站点的物种丰富度,包括Chao1 (图5-a)、ACE (图5-b) 和 Observed species指数 (图5-c) 均极显著高于D3、D8、F3和F8站点 (P<0.01),尤以H8和I9最高。各站点的群落多样性,包括Simpson(图5-d) 和 Shannon 指数 (图5-e),也表现出了显著差异 (P<0.05),其中D3站点最高,F3站点最低。然而,基于宿主目水平的分组结果显示,不同物种分组间的Alpha指数均无显著差异 (P>0.05,图5-f,以Chao1指数为例)。上述结果表明,不同站点细菌群落多样性和丰富度指数均存在显著差异,而基于宿主目水平的分组之间未表现出显著差异。
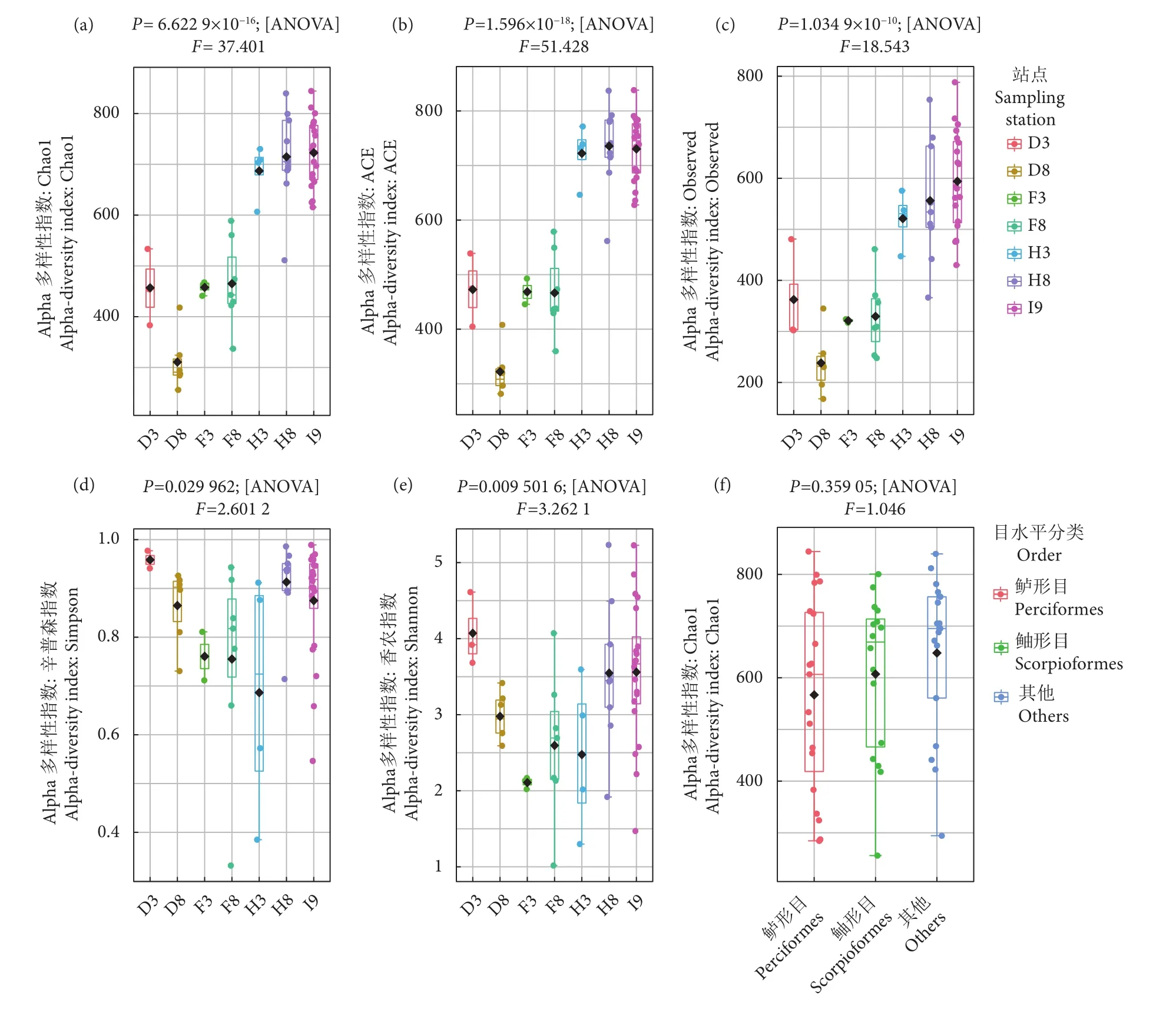
图5 Alpha多样性指数箱线图注:a—e. 基于站点分组的Chao1、ACE、Observed、Simpson、Shannon 多样性指数箱线图,不同颜色代表不同站点的样本;f. 基于样本宿主(目水平) 分类分组的Chao1 指数箱线图。Fig. 5 Boxplot of Alpha diversity indexNote:a—e. Boxplot of diversity indexes of Chao1, ACE, Observed, Simpson and Shannon based on site grouping, and different colors represent samples of different sites; f. Boxplot of Chao1 index based on sample host (Order level) classification.
2.4 Beta 多样性分析
Beta多样性可用于描述不同样本微生物群落之间物种组成的差异。我们基于不同分组对各个样本进行NMDS分析 (图6)。按照站点分组结果显示,各个站点样本的Beta多样性具有极显著差异(P<0.001),且不同站点样本分别聚集在一起 (图6-a)。总体来看,与α多样性结果一致,即H3、H8、I9站点与D3、D8、F3、F8站点明显分为两个主要类群。其中D3和F3、D8 和F8 站点的样本距离更近,H3、H8和I9站点的10个样本聚集在一起,I9站点一部分样本单独分群(图6-a:I9-2)。然而,基于样本目水平的分组未发现明显分群,不同的目分组间不存在显著差异 (P>0.05,图6-b)。由此可见,地理站位对硬骨鱼鳃组织内微生物可能具有重要影响,而样本宿主遗传因素可能不是影响鱼鳃细菌群落分布的主要因素。
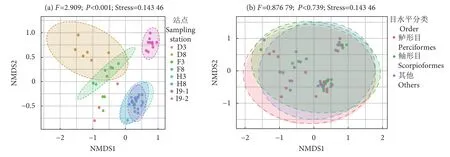
图6 基于不同分组方式的NMDS分析 [非参数多元方差分析 (PerMANOVA)]注:a. 基于站点分组的 NMDS 分析; b. 基于样本的目水平分组。图中两个样本点越接近,表示两个样本物种组成越相似。横纵坐标表示样本间的相对距离,无实际意义。图形中的每一个点代表一个样本,不同颜色代表不同的样本分组信息。NMDS 结果的优劣用胁迫系数(stress) 来衡量,此值越小越好,当小于 0.2 时表示可以用 NMDS 的二维点图表示组间或组内差异。Fig. 6 NMDS analysis based on different grouping modes [Nonparametric multivariate analysis of variance (PerMANOVA)]Note:a. NMDS analysis based on site grouping; b. Grouping based on the order level of the sample. The closer the two sample points are,the more similar the composition of the two sample species are. The abscissa and ordinate indicate the relative distance between samples, which is of no practical significance. Each point in the graph represents a sample, and different colors represent different sample grouping information. The advantages and disadvantages of testing NMDS results are measured by stress coefficients, the smaller, the better, and the value less than 0.2 indicates that the twodimensional dot plot of NMDS can be used to represent differences between or within groups.
2.5 微生物群落功能预测
基于KEGG比对共获得8个一级层级和41个二级层级功能分类。各样本在一级功能层级组成上的差异不明显 (数据未展示),占比最高的均为基因与新陈代谢相关功能 (Metabolism, 50%),其次是遗传信息处理 (Genetic information processing, 16%)、环境信息处理 (Environmental information processing, 13%)。图7为KEGG数据库比对到的22个相对丰度较高 (>1%) 的二级功能层级热图。总体来看,二级功能层级中膜转运 (Membrane transport)功能丰度最高 (11.02%),其次为氨基酸代谢(Amino acid metabolism, 10.5%)。但不同功能在不同样本中丰度差异明显,膜转运 (14%)、碳水化合物代谢 (Carbohydrate metabolism, 10%)、氨基酸代谢 (9%)、复制和修复 (Replication and repair, 9%)在I9.11S中丰度最高,在D8.6S中次之。样本聚类显示鱼鳃中细菌功能基因分布及丰度与站点没有明显的关联性。另外,基于COG数据库预测结果与KEGG的一级功能结果类似 (数据未展示),各样本中的功能基因比例总体差别不大。
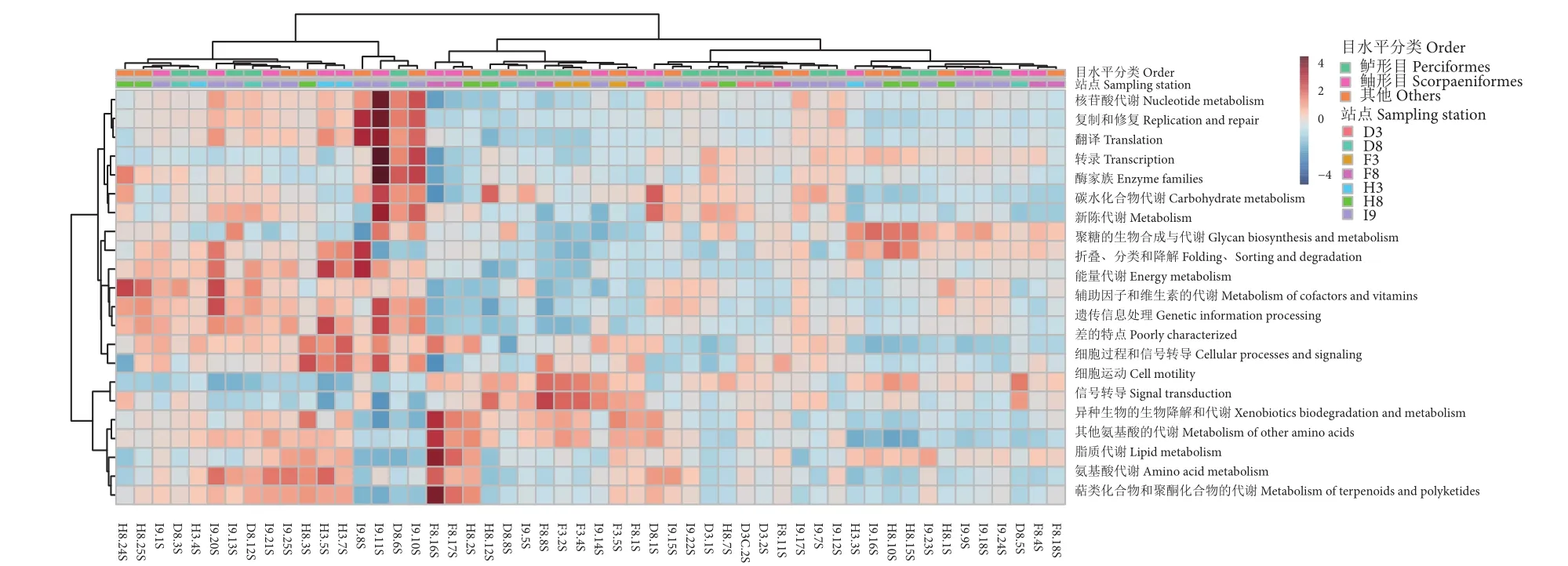
图7 22个KEGG-L2功能层级相对丰度热图注:横坐标代表不同的样本,纵坐标代表不同的功能层级,图片上方色条表示不同分组,并按照样本聚类。Fig. 7 Heatmap of relative abundance of 22 KEGG-L2 functional levelsNote:The abscissa represents different samples; the ordinate represents different functional levels; the color bars indicate different groups, which are clustered according to samples.
两种功能数据库预测结果均表明,鱼鳃细菌群落可能主要参与了宿主营养物质转运及代谢等相关进程。
3 讨论
3.1 海洋鱼类鳃组织菌群群落组成
本研究发现,硬骨鱼鳃组织细菌群落中的优势菌门包括变形菌门、厚壁菌门、浮霉菌门、放线菌门、拟杆菌门和蓝细菌,这与其他学者对海洋环境菌群统计分析所得结果一致,如穆大帅等[24]对我国分离鉴定的577种海洋细菌进行多样性分析,发现在门分类水平上分布最多的是变形菌门,其次是拟杆菌门、放线菌门和厚壁菌门。白洁等[25]采用高通量测序方法分析了南海南部海域浮游细菌丰度,发现南海南部海域浮游细菌的优势类群为变形菌门、蓝藻门和拟杆菌门,优势亚群为γ-变形菌纲、α-变形菌纲、蓝藻菌纲和黄杆菌纲。徐新亚等[26]报道广西北部湾分离鉴定到1 843种海洋细菌,分别隶属于放线菌门、拟杆菌门、厚壁菌门、浮霉菌门和变形菌门。本研究结果表明海洋环境与鱼类鳃中的微生物群落至少在门水平上具有较高的一致性。变形菌门作为海洋中主要的微生物类群之一,在海洋生态系统的碳循环、氮循环等多种物质循环过程中均发挥重要作用[27],有研究证实海洋表面的变形菌门含有丰富的固氮种群[28]。厚壁菌门最主要的功能是降解纤维类物质,拟杆菌门主要降解非纤维物质[29-30],而上述3类细菌在鱼鳃组织上的富集可能与宿主在海洋环境中的代谢需求有关。此外,Legrand等[31]认为,黄尾鰤 (Seriolalalandi) 鳃中丰度较高的浮霉菌门可能在海洋鱼类体内固氮作用中发挥功能,有助于促进氮元素在海洋环境与海洋生物之间的垂直传播。由于本研究是通过16S的物种组成反推预测群落功能,并未直接对宏基因组和功能基因进行测序。因此,功能分析结果会与真实情况有一定差距。未来研究还需进一步结合宏基因组测序及海洋环境有机物质的测定,才能更好地解读鳃组织微生物在鱼体环境适应性方面发挥的功能。
3.2 海洋鱼类鳃组织菌群的潜在生态功能
近年来,国内外学者在海洋细菌多样性研究领域贡献了大量成果,但多数只关注水体中游离的细菌,对于宿主,尤其是鳃组织上,共附生细菌群落潜在功能的研究相对较少。本研究预测到鱼类鳃中细菌多参与宿主的新陈代谢过程,积极参与氨基酸代谢反应和碳水化合物代谢反应,表明鱼鳃中细菌可能在碳循环中发挥作用。Kessel等[32]研究了鲤(Cyprinus carpio) 和斑马鱼 (Danio rerio) 鳃中微生物在氮循环中的作用,结果发现鳃中含有一个活跃的N-循环细菌群,属于β-变形杆菌纲成员,可能对许多鱼类降低氨浓度、防止高浓度氨中毒有益。本研究发现所有样本中均包含丰度不一的β-变形菌纲成员,提示海水鱼类鳃组织中可能也具有类似功能的菌群。Pratte等[14]对于法国Moorea珊瑚礁采集的鱼鳃和肠道等样本的微生物群落进行研究,发现成鱼和幼鱼鳃中的微生物群落组成与肠道有显著差异。鳃中最丰富的细菌是希瓦氏菌属和Endozoicimonaceae成员,且微生物群落与环境样品共享的OTU百分比高达75%,表明鳃微生物组可能受到周围浮游微生物群落变化的影响。本研究中D8 站点的网纹裸胸鳝 (Gymnothorax reticularis)、黑鮟鱇 (Lophiomus setigerus)、环纹蓑鲥 (Pterois lunulata) 和深水金线鱼 (Nemipterus bathybius) 中的希瓦氏菌属细菌占比同样较高,与该结果吻合。然而,其他多数样品中则是变形菌门的不动杆菌属细菌比例较高,是所有站点的优势菌属。该结果说明不同站点或不同宿主物种的微生物组成可能差异很大。有研究报道,不动杆菌属细菌作为非发酵革兰阴性菌,具有代谢多样性,可有效降解海水中己内酰胺、石油油烃等海洋污染成分[33]。不动杆菌属在海水鱼鳃部的富集或可有效促进环境污染物等的代谢,从而对宿主产生潜在的保护作用。除了D3站点,其他6个站点的优势菌属还包括嗜冷杆菌属。该属细菌多分布于潮湿、含盐或寒冷的生境中,且能分泌适应低温的酶,可在海洋低温下发挥其生物活性[34],且具有较高的催化效率。然而,本研究发现的广泛存在于鱼鳃组织中的嗜冷杆菌是否也具有类似功能值得深入研究。
3.3 地理位置对鱼鳃细菌群落的影响
本研究发现站点对鱼鳃细菌群落的影响远高于宿主物种遗传因素的影响。近年来,其他研究者也发现地理位置对海洋环境中的微生物群落产生影响。翟万营等[35]发现地理位置对南极中山站附近海水微生物群落组成有重要影响;Zhang等[36]认为除理化性质外,地理位置对南极中山站、长城站周边土壤样品和近海沉积物中的细菌群落组成产生一定影响。与土壤等环境样本不同,生物体内的微生物群落除受到环境影响外,还会受到饮食、遗传、发育阶段、健康等因素的影响。本研究发现海洋鱼类鳃中的菌群也受到地理位置的影响,可能由不同站点的水质环境、浮游微生物和营养物质与鳃的直接接触造成。海洋动物鳃组织与肠道等内生组织不同,鳃暴露于水体环境中,没有额外的免疫屏障,可能导致鱼鳃细菌群落受到环境的影响尤为显著。鉴于海洋鱼类具有自主、灵活的游泳能力,由此推测,其在海洋微生物传播和海洋微生物群落调节中可能发挥着重要作用。
此外,本研究还发现I9站点的20个样本明显分为两个群,这可能有两个原因:1) 两个群落在宿主物种水平上存在差异,第一个群落包括大鳞鳞头鲉 (Sebastapistes megalepis)、黄纹拟鲈 (Parapercis xanthozona)、寿鱼 (Banjos banjos) 和棕斑宽吻鲀(Amblyrhynchotes rufopunctatus) 等物种;第二个群落包括冠鲽 (Samaris cristatus)、叉斑狗母鱼 (Synodus macrops)、美尾䲗 (Calliurichthys japonicus)、虻鲉 (Erisphex pottii) 和红鲬 (Bembras japonicus) 等物种。通过调查采样记录发现,第一个类群部分物种鱼类营养级高于第二个类群,而第二个类群的样本鱼体较小,由此推测,营养级和个体大小 (发育阶段) 或许是造成该批次群落差异的一个原因。2) 扩增、建库、测序等试验环节批次的不同也有可能会对上述群落差异造成一定影响,具体原因还需要更多试验证实。
综上所述,南海具有丰富的海洋微生物资源,本研究发现南海近岸硬骨鱼鳃组织中细菌群落的组成非常丰富。采样站点相比宿主来源对鳃组织上细菌群落结构及多样性具有更主要的影响,它们可能在辅助宿主营养物质转运及代谢方面发挥积极作用。然而本研究也存在很多不足如:样本和站点数量较少,未对海水样品进行对照研究等。未来还需进一步扩大样品的种类、数量,并结合水质环境、理化因子和宿主的生理生长指标及生活方式等多维度指标,同时探讨海洋动物鳃、肠道等多个器官中海洋微生物的分布、富集和传播模式,为研究海洋动物宿主微生物与环境浮游微生物群落之间的互作关系及生态影响提供更深入、全面的数据支持。
猜你喜欢
中国农学通报(2022年14期)2022-06-01
油气田环境保护(2022年2期)2022-05-09
山东畜牧兽医(2021年5期)2021-06-07
科学(2020年3期)2020-11-26
儿童时代·幸福宝宝(2020年9期)2020-09-08
当代水产(2020年3期)2020-06-15
中国沼气(2019年1期)2019-04-13
探索科学(2017年5期)2017-06-21
小星星·阅读100分(高年级)(2015年11期)2015-11-28
医学研究杂志(2015年12期)2015-06-10
