
RNA测序揭示肾母细胞瘤中潜在的circRNA-miRNA-mRNA调控网络
2022-10-10 05:50田小毛金黎明陈美玲魏光辉400014重庆重庆医科大学附属儿童医院泌尿外科儿童泌尿生殖发育与组织工程重点实验室儿童发育疾病研究教育部重点实验室国家儿童健康与疾病临床医学研究中心儿童发育重大疾病国家国际科技合作基地儿科学重庆市重点实验室
第三军医大学学报 2022年19期
向 彬,田小毛,米 滔,金黎明, 陈美玲, 刘 丰,刘 星,林 涛,魏光辉 400014 重庆,重庆医科大学附属儿童医院泌尿外科,儿童泌尿生殖发育与组织工程重点实验室,儿童发育疾病研究教育部重点实验室,国家儿童健康与疾病临床医学研究中心,儿童发育重大疾病国家国际科技合作基地,儿科学重庆市重点实验室
肾母细胞瘤(wilms tumor,WT)是最常见的儿童肾脏恶性肿瘤,在所有儿童恶性肿瘤中约占6%;15岁以下儿童中,发病率约为7.1/100 000[1]。随着化疗、手术、放疗等多种治疗手段的综合应用,总体生存率已经超过90%[2]。然而,间变病理类型和复发患者的预后仍然很差[3-5]。进一步探索WT发生发展的分子机制对WT患者接受更好的治疗、获得更好的整体预后具有重要意义。环状RNA(circRNA)是一类新型的非编码RNA,其主要定位于细胞质中,提示它们在转录后水平发挥调控作用[6]。然而,circRNA表达谱及其在肾母细胞瘤发生发展中的作用机制仍不清楚。既往的研究表明circRNAs调控肿瘤进展的主要途径是作为一种竞争性内源性RNA(ceRNA),通过miRNA的海绵作用进一步调节下游mRNA的表达[7-8]。因此,为了探索circRNA相关的ceRNA调控网络在肾母细胞瘤发生发展过程中的潜在机制,本研究对临床来源的肿瘤样本进行测序分析,鉴定差异表达的circRNAs和mRNAs;并基于自己的测序数据构建WT中一个潜在的circRNA-miRNA-mRNA调控网络。
1 材料与方法
1.1 肿瘤组织样本
本研究方案获得重庆医科大学附属儿童医院伦理委员会批准(批件号:2022年伦审第50号)。收集2021年6月至2022年3月期间重庆医科大学附属儿童医院泌尿外科的WT患者的肿瘤切除组织。根据美国儿童肿瘤协作组(Children’s Oncology Group,COG)标准[9](没有前期化疗),所有样本诊断为肾母细胞瘤。如果病理结果报告神经母细胞瘤、横纹肌样瘤或透明细胞肉瘤等,则被排除。共纳入25例配对的肿瘤及邻近正常组织标本。选择8对新鲜的肿瘤和配对正常组织进行mRNA测序,获得肾母细胞瘤中mRNA的表达数据;选择4对配对的肿瘤组织进行circRNA测序,以获得完整的circRNA表达数据。剩余13对配对的肿瘤标本被冷冻储存在液氮中,用于RT-qPCR验证。
1.2 circRNA测序和mRNA测序
按照提示,用RiboZero rRNA去除试剂盒(Epicentre,WI,USA)处理4对WT样品的总RNA。将rRNA耗尽和RNase R酶切后的RNA样品进行片段化,用随机引物合成cDNA。纯化cDNA的PCR扩增产物,用NovaSeq6000(Illumina,San Diego,CA,USA)对RNA文库进行质量控制和测序。使用fastp[10]软件对序列进行过滤,获得可用于数据分析的“clean reads”。在得到测序读数以后,采用CIRI软件[11]对circRNA进行预测。在以上预测的基础上,根据circRNA在染色体上的位置信息,合并全部样本中的circRNA结果对合并后结果进行重新编码ID。而后将其与circBase数据库[12]进行比对,从而识别出已注释的和新预测的circRNA。
本次mRNA测序为16个样本的双端测序。使用RNeasy mini试剂盒(Qiagen, Germany)提取总RNA。使用TruSeqTMRNA样品制备试剂盒(Illumina,USA)按照指南合成双端文库。纯化的文库用Qubit® 2.0荧光仪(Life Technologies,USA)进行定量,并由Agilent 2100生物分析仪(Agilent Technologies, USA)进行验证,以确认插入物的大小并计算出分子浓度。在Illumina NovaSeq 6000上进行测序。使用FastQC软件对测序得到的结果进行质量评估。使用FASTX-Toolkit软件(http://hannonlab. cshl.edu/fastx_toolkit/)对原始序列进行清洗过滤得到“clean reads”。使用Hisat2软件[13]将得到的“clean reads”比对到已知的参考基因组(GRCh38.91)上。
1.3 circRNA和mRNA的差异表达分析
为了量化mRNA的表达丰度,使用FPKM(Fragments PerKilobase Million)来表征不同基因的表达量,其计算公式如下:
首先使用Stringtie软件[14]对比对后每个基因区段内的片段进行计数,然后再使用TMM(trimmed mean of M values)算法进行归一化,最后再计算每个基因的FPKM值。在得到基因的FPKM表达值以后,使用edgeR软件包[15]对组间基因表达进行差异分析(配对t检验),得出组间基因表达的P值以及经过多重比较检验后校正的Q值。同时,根据FPKM值计算差异表达倍数(fold change,FC)。DEmRNAs的筛选标准为Q<0.05和|logFC|>2。
无论是新预测的还是已被数据库注释的circRNA,目前绝大部分的circRNA都无法获得其完整的全长序列。只能使用circRNA序列上的“back-splicing”(反向剪切)位点部分的“junction reads”的数量来作为circRNA表达量的计算依据。通常使用SRPBM(spliced reads per billion mapping)值来表征circRNA的表达水平[16], 其计算公式如下:
差异表达分析与mRNA分析策略一样,也使用edgeR来计算。DEcircRNAs的筛选标准为|FC (fold change)|>2和P<0.05。
1.4 基因本体论(gene ontology,GO)和京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)通路富集分析
根据circRNA在基因组上的位置信息,可以获得与circRNA所在基因组位置上对应的蛋白编码基因的信息,这类基因被称为某一circRNA的“parental gene”(亲本基因)。为了探索circRNA相关ceRNA网络的潜在生物学机制,使用R“ClusterProfiler”软件包[17]对DEcircRNA的亲本基因和DEmRNA进行基于Gene Ontology,包括生物学过程(biological process,BP)、细胞成分(cellular component,CC)、分子功能(molecular function,MF)和KEGG途径的富集分析。P值<0.05被认为具有统计学意义。
1.5 构建circRNA-miRNA-mRNA调控网络
ceRNA假说揭示了一种RNA间相互作用的新机制。mRNA和circRNA可以通过竞争性的结合miRNA来调控彼此的表达。miRanda是一种常用于检测基因组序列中潜在microRNA靶位点的算法[18]。匹配策略主要包括两个步骤:首先,在查询的miRNA序列和基因3’UTR序列间进行动态规划局部比对,基于序列互补性(而不是序列一致性)打分;然后,基于第一步获得的高分比对结果(sc参数控制),使用RNAlib(来自ViennaRNA包)评估比对的热力学稳定性。最终能量小于阈值的匹配对象作为最终结果。在前面的结果中,获得了差异表达的circRNA和mRNA。使用miRanda鉴定差异circRNA和mRNA的microRNA靶点,初步匹配条件为序列互补Tot Score>140和热力学稳定性Tot Energy<-20。然后筛选出与miRNAs共调控的DEmRNA和DEcircRNA。利用miRanda回归模型分析以及序列匹配的方法(筛选条件设置为Sum max energy<-90)建立miRNAs的海绵吸附作用的调控网络。最后根据DEcircRNAs与对应DEmRNAs的协同表达来找到核心的ceRNA 网络。并用Cytoscape软件v.3.8.2[19]进行可视化。
1.6 基因集富集分析(Gene Set Enrichment Analysis,GSEA)
GSEA用于确定预先定义的基因集合在两种生物状态(如肿瘤和正常组织)之间是否显示出统计学差异[20]。基于“Molecular Signatures Database v7.4”,利用GSEA分析差异基因的潜在途径。基于“curated gene sets”确定丰富的KEGG途径、生物学过程、细胞成分和分子功能。最后,基于“immunologic signature gene sets”确定与肿瘤系统中细胞活性和应答相关的特定生物学状态或过程。根据默认的加权富集统计方法对数据进行归一化,以获得归一化富集得分(NES)。|NES|>2和P<0.05的基因集被认为具有统计学意义。
1.7 蛋白互作网络(Protein-Protein Interaction Networks,PPI)的构建和HUB基因的鉴定
使用中等置信度(0.400)的交互得分,利用在线网站STRING[21-22]构建了一个基于ceRNA调控网络中靶基因的蛋白质-蛋白质相互作用网络。使用Cytoscape软件对PPI网络进行可视化。随后,使用cytoHubba 插件[23]识别前10个HUB基因。
1.8 基于HUB基因的预后价值构建关键的circRNA调控子网络
TARGET数据库是目前最大的儿童肿瘤综合数据库。本研究从中下载了136个WT样本的RNA测序数据和相应的临床信息数据集。在生存结果分析中,根据HUB基因表达值的中位数将患者分为高表达组和低表达组。选择无病生存率(disease free survival,DFS)作为主要随访终点,使用Kaplan-Meier方法和基于TARGET数据集表达谱的单因素Cox回归分析来评估HUB基因的预后价值。最后,基于关键的HUB基因构建WT中的circRNA调控子网络。
1.9 RNA测序结果的PCR验证
使用Trizol试剂(Invitrogen, Carlsbad, CA, USA)提取每个肿瘤组织的RNA。根据制造商的说明,使用Simply P Total RNA Extraction Kit(BioFlux, China)提取细胞总RNA。使用NanodropOne(Thermo Fisher Scientific,Waltham,MA,United States)测定RNA浓度。使用PrimeScriptTMRT试剂盒(TaKaRa, Japan)对circRNA和mRNA进行反转录,并使用TB Green® PremixExTaqTMⅡ试剂盒(TaKaRa,Japan)进行扩增和定量。实时PCR在CFX Connect Real-Time PCR Detection System上操作。每个样本包含3个技术重复。GAPDH用于circRNA和mRNA表达的标准化。目标基因的表达基于2-ΔΔCt公式计算。本研究使用的引物见表1。
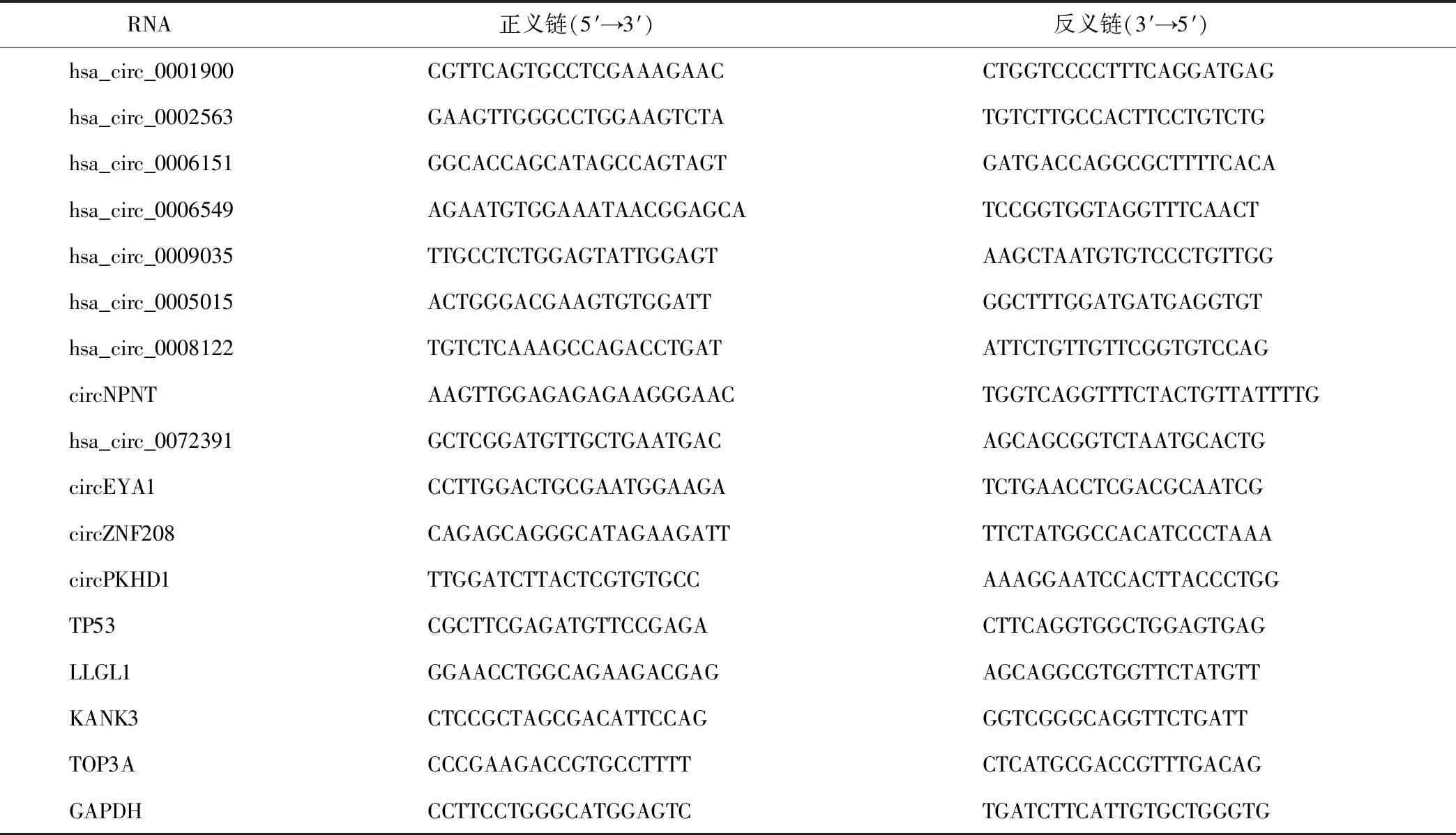
表1 RT-qPCR引物
1.10 功能丧失实验
本研究选择hsa_circ_0009035在WT细胞系中进一步验证功能。针对hsa_circ_0009035后剪接位点的siRNA和阴性对照由Hanbio(上海,中国)设计并合成。使用脂质体2000 (Invitrogen,美国)进行细胞转染。根据制造商的说明步骤,采用CCK-8法(MCE,HY-K0301)检测细胞活力,并采用划痕法检测细胞迁移。使用基质凝胶(Biozellen,B-P-00002-4,中国)敷在24孔板小室(Falcon,353097,美国)中进行Transwell检测细胞侵袭。使用BD细胞周期检测试剂盒和流式细胞仪检测细胞周期时相分布。
1.11 数据分析
配对组织样本的基因表达差异分析采用配对t检验(正态分布)或Wilcoxon配对检验(非正态分布)。使用GraphPad Prism 8对PCR实验结果进行分析。 所有的生物信息学分析使用R软件4.0.3版进行。检验水准α=0.05。
2 结果
2.1 WT中差异表达的circRNA和mRNA鉴定
对4对肾母细胞瘤和配对正常组织样本进行circRNA测序,共鉴定出23 978个circRNA,其中10 884个circRNA在circBase(http://www.circbase.org/)数据库中已获得注释(图1A)。另外,结果显示大多数circRNA(84.18%)由外显子组成,11.72%由内含子组成,而4.10%定位于基因间区(图1B)。这里排除了来自基因间区的circRNA(目前认为该类circRNA无功能)。通过热图显示了筛选到的DEcircRNA的特异性表达(图1C),层次聚类分析显示在WT中大多数DEcircRNA低表达。DEcircRNA的分布通过火山图显示(图1E),总共鉴定出314个DEcircRNA,其中115个上调、199个下调。同样,本研究额外对8对配对的肾母细胞瘤组织进行了测序,分析了mRNA表达谱。根据筛选标准,检测到1 030个上调和582个下调差异表达基因。图1D展示了肿瘤和正常组织的基因表达热图,图1F展示了差异表达基因的火山图。
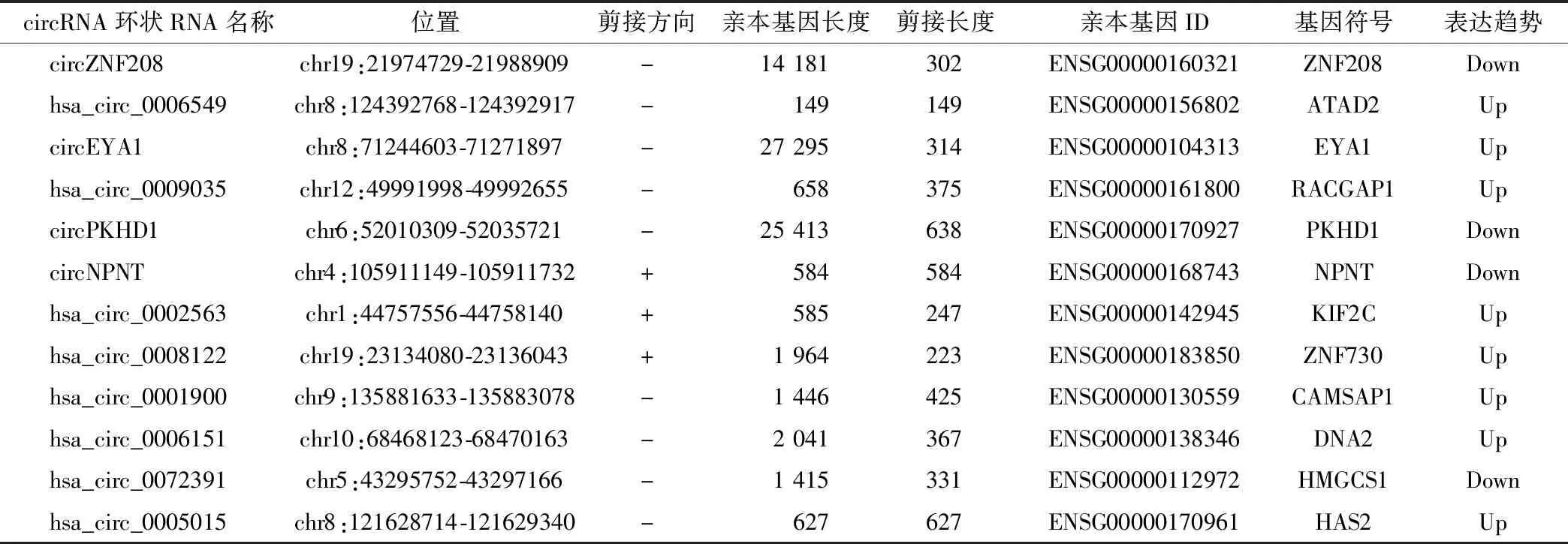
表2 拟PCR验证的DEcircRNA的基本特征
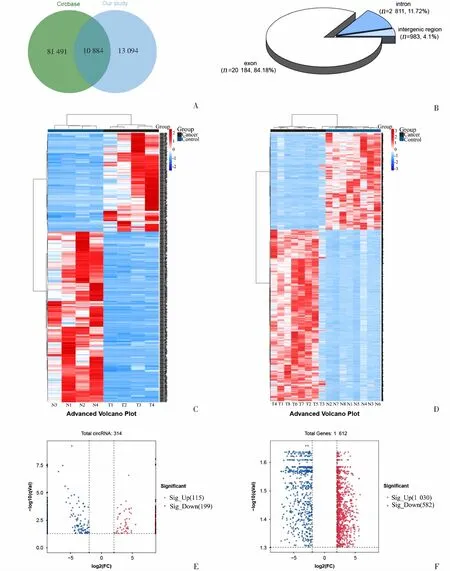
图1 基于RNA高通量测序分析WT中差异表达circRNA和mRNA
为了验证circRNA测序结果,随机选择了12个DEcircRNA(8个上调,4个下调)设计了跨越circRNA后剪接位点的特异性发散引物进行组织验证(DEcircRNA的详细特征见表2)。RT-qPCR结果显示其中7个DEcircRNA显示出显著的表达差异(图2)。
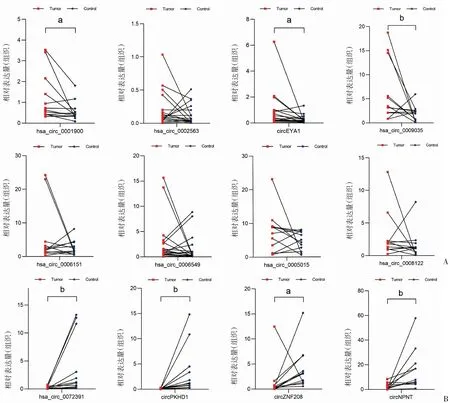
a:P<0.05,b:P<0.01;红点代表肿瘤样本,蓝点代表正常样本
2.2 DEmRNA和DEcircRNA亲本基因的GO和KEGG途径富集分析
为了发现DEcircRNA和DEmRNA的潜在功能,对DEcircRNA亲本基因和DEmRNA分别进行GO注释和KEGG通路富集分析。DEcircRNA亲本基因GO和KEGG通路富集分析结果如图3A、C。GO注释分析表明DEcircRNA亲本基因主要定位于细胞质和外泌体,参与信号转导、细胞增殖和细胞周期等过程。KEGG途径富集分析表明,DEcircRNA亲本基因主要富集在多条癌症相关的途径中。此外,本研究对DEmRNA同样进行基于GO和KEGG的富集分析。对于GO注释(图3B),与恶性表型相关的生物学过程显著丰富,包括DNA复制、细胞分裂、细胞周期和细胞增殖等过程。KEGG途径(图3D)富集分析表明,DEmRNA主要集中在细胞增殖、代谢和生长的信号通路上,另外肿瘤相关信号通路如P53信号通路和多条代谢通路也得到了富集。
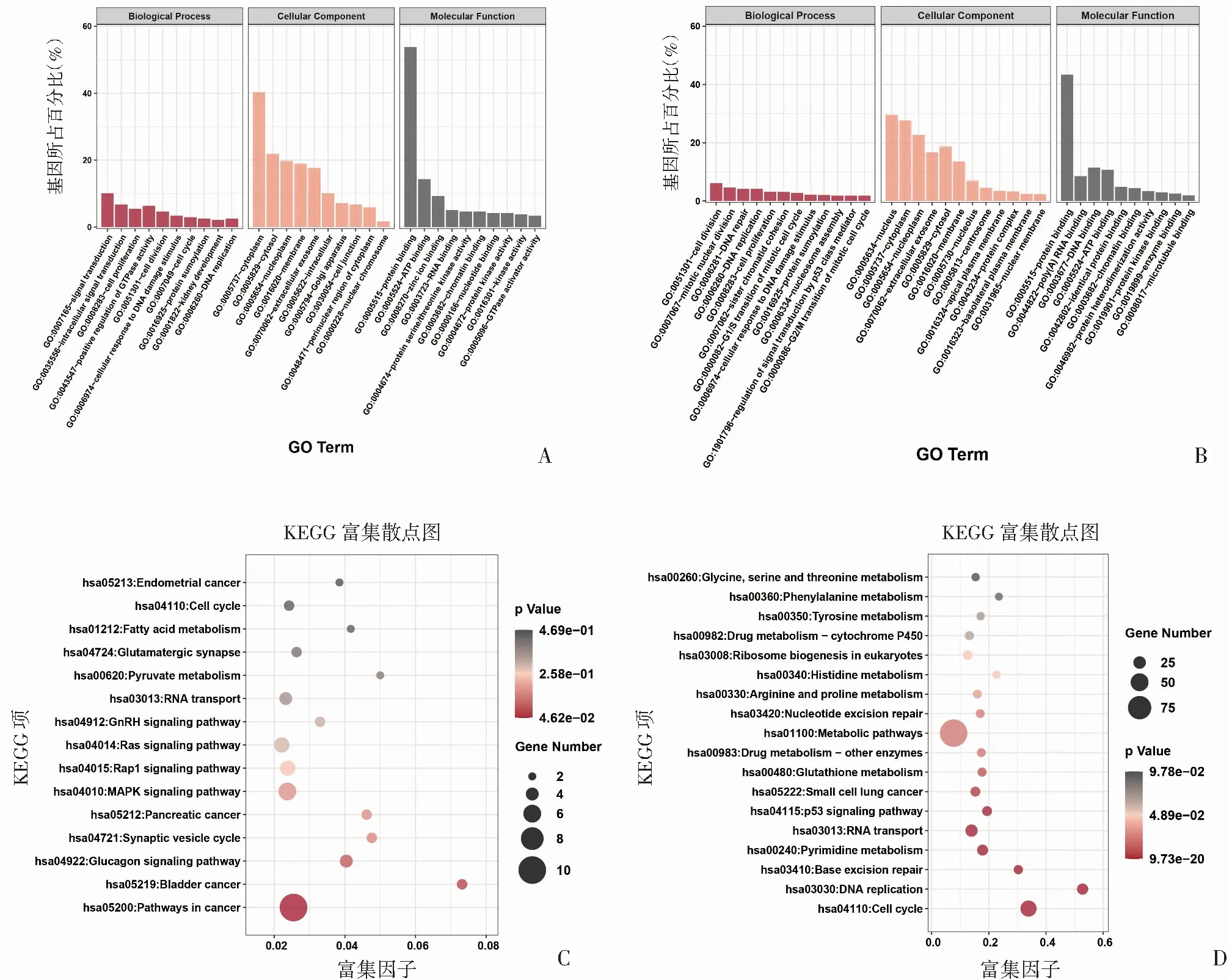
A:DEcircRNA亲本基因的GO富集分析;B:DEmRNA的GO富集分析;C:DEcircRNA亲本基因的KEGG富集分析;D:DEmRNA的KEGG富集分析
2.3 WT中circRNA-miRNA-mRNA调控网络的构建
为了预测所有DEcircRNA和DEmRNA的潜在靶点miRNA,使用miRanda预测工具分别识别314个DEcircRNA和1 612个DEmRNA的miRNA靶点。构建了DEcircRNA-miRNA和DEmRNA-miRNA相互作用网络(图4A、B)。通过整合circRNA-miRNA对和miRNA-mRNA对,基于相同miRNA结合位点构建了一个完整的circRNA-miRNA-mRNA网络。根据ceRNA理论,本研究选择circRNA和mRNA具有协同表达趋势的ceRNA网络,并将筛选条件设置为Sum max energy<-90(绝对值越大越严格)。最终,构建了一个精确的circRNA-miRNA-mRNA调控网络,其中包括62个circRNA、8个miRNA和127个mRNA,并随后通过Cytoscape进行可视化(图4C、D)。
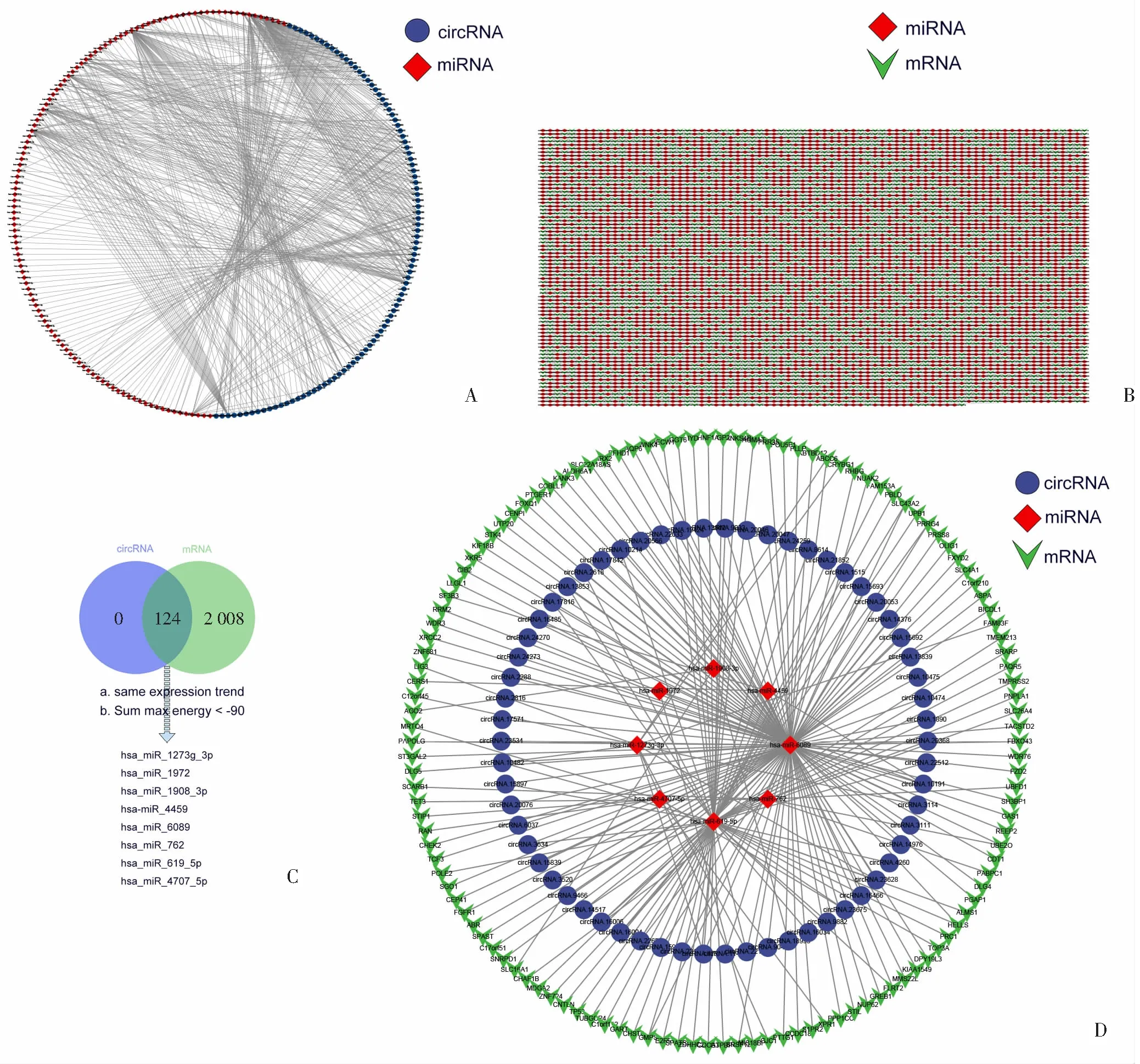
A:circRNA-miRNA调控网络;B:miRNA-mRNA调控网络;C:circRNA和mRNA的miRNA靶点交集;D:ceRNA调控网络
2.4 ceRNA网络中靶基因的GSEA富集和PPI网络分析
为了研究ceRNA调控网络在WT中的潜在生物学功能,基于所涉及的127个DEmRNA进行GSEA分析。结果发现DEmRNA的富集通路见图5A,尤其是主要与细胞周期相关。值得注意的是,基于免疫背景基因集的富集分析显示DEmRNA与多种免疫细胞的活性有关(图5B)。结果表明,这些DEmRNA在一定程度上参与了肿瘤的发生发展,特别是与细胞周期和免疫应答有关。为了探索关键的HUB基因,基于STRING数据库建立了包含127个点和170个连接的PPI网络(图5C)。前10个HUB基因,包括TP53、KANK3、STIL、RRM2、HELLS、POLE2、TOP3A、LLGL1、GMPS和GTPBP4被鉴定(图5D)。
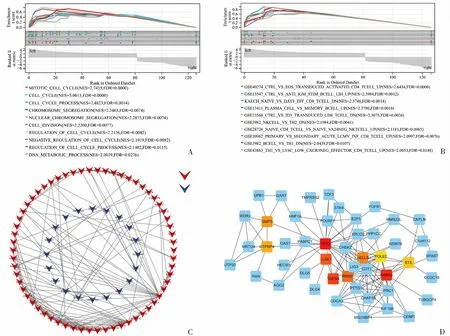
A:基于C5策划基因集对ceRNA网络中DEmRNA的GSEA分析;B:基于免疫特征基因集对ceRNA网络中DEmRNA的GSEA分析;C:ceRNA网络中DEmRNA的PPI网络;D:通过cytoHubba插件鉴定前10个HUB基因
2.5 基于HUB基因的预后特征构建关键的ceRNA调控子网络
在鉴定的前10个HUB基因中,除KANK3外,其余HUB基因都呈表达上调趋势(图6A)。为验证HUB基因的预后价值,通过单因素COX回归分析10个HUB基因对WT患者远期生存率的影响(图6B)。结果显示4个HUB基因包括TP53、KANK3、LLGL1和TOP3A的mRNA表达水平与WT患者的RFS(无病生存率)显著相关(图6C)。结果提示这4个基因可能在WT的进展和预后中起关键作用。最后,基于这4个HUB基因的重要性,进一步构建了靶向该基因的子网络(图6D)。RT-qPCR用于验证4个HUB基因的表达水平,结果显示出与测序数据一致的趋势(图6E)。
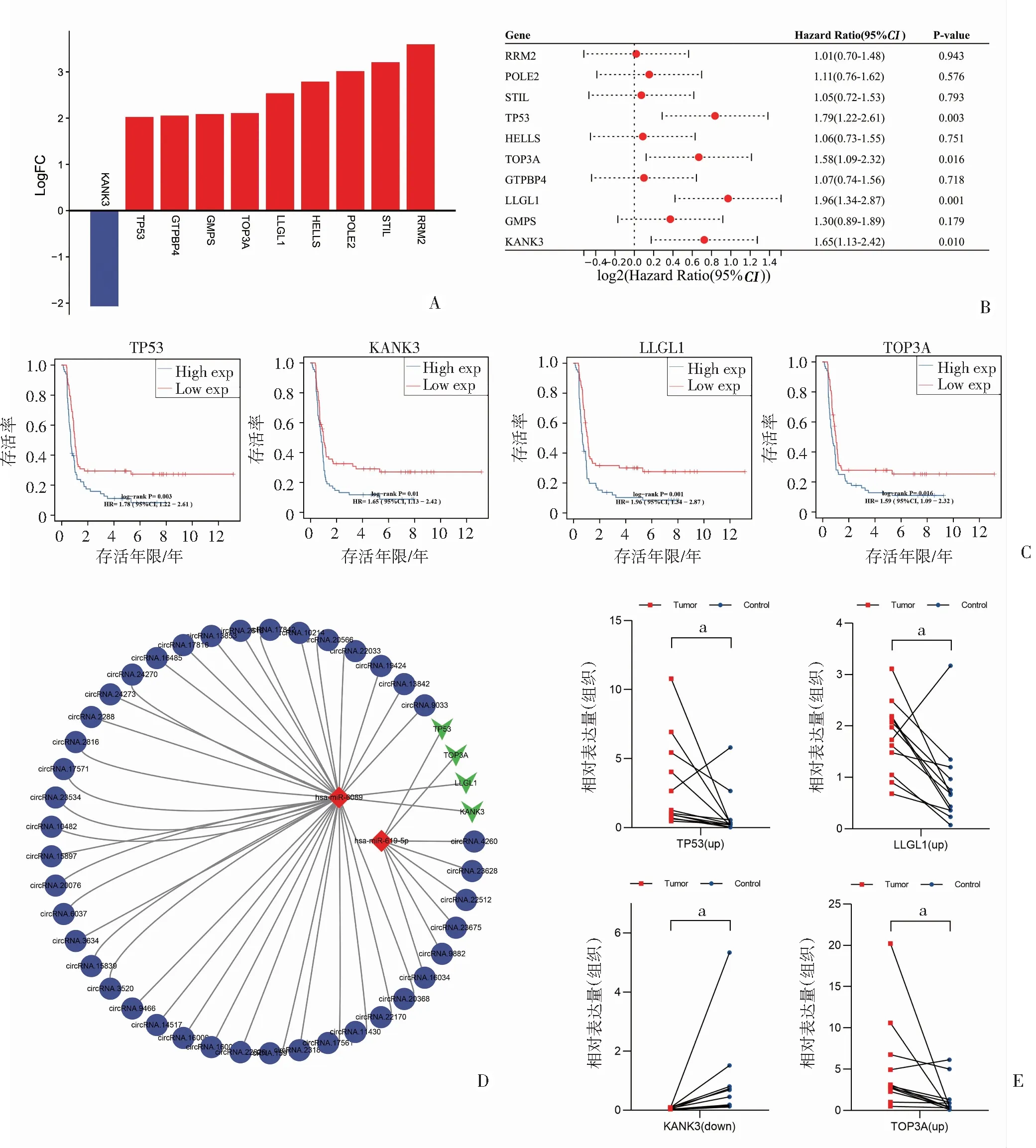
A:前10个HUB基因表达变化柱状图(红色柱代表上调基因,蓝色柱代表下调基因);B:森林图显示单因素Cox回归分析中HUB基因的mRNA表达水平与WT患者预后的关联;C:与WT患者预后显著相关的4个HUB基因的生存曲线;D:基于4个与预后显著相关的HUB基因构建ceRNA调控子网络;E:对关键HUB基因的RT-qPCR验证 红点代表肿瘤样本,蓝色代表正常样本;a:P<0.01
2.6 沉默hsa_circ_0009035抑制WT细胞的增殖、侵袭和迁移
本研究选择子网络中的hsa_circ_0009035进行进一步的功能验证。如前所述,与邻近的非肿瘤组织相比,hsa_circ_0009035在WT组织中高表达。PCR产物的Sanger测序验证了其反向剪接位点(图7A)。为了研究hsa_circ_0009035在WT进展中的作用,本研究设计了特异性沉默hsa_circ_0009035的siRNA。siRNA片段通过靶向hsa_circ_0009035的反向剪接位点敲低目的环状RNA的表达水平。与阴性对照相比,hsa_circ_0009035的沉默显著降低了WIT-49细胞中的RNA水平(图7B)。通过CCK-8实验,沉默hsa_circ_0009035显著抑制了WT细胞的增殖能力(图7C)。此外,沉默hsa_circ_0009035后,细胞的迁移和侵袭能力下降(图7D、E)。细胞周期分布分析显示,hsa_circ_0009035沉默后,G1期细胞比例明显减少,S期细胞比例增加(图7F)。以上结果表明构建的环状RNA网络在WT进展中具有潜在作用。
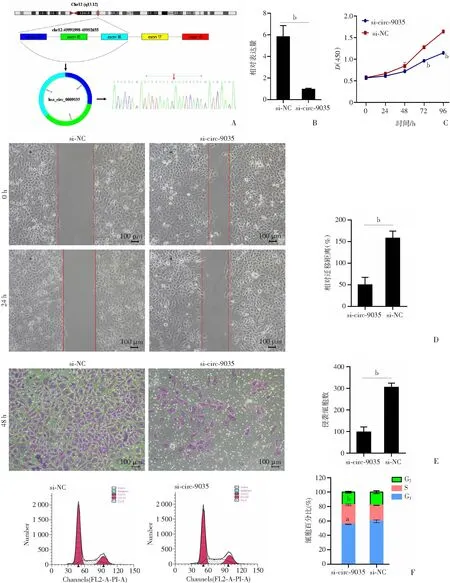
a:P<0.05,b:P<0.01
3 讨论
当前对WT的治疗已取得进展,但影响其发生和发展的具体机制仍不明确,特别是在circRNA领域。因此,探讨WT发生发展的分子机制,并识别有效的生物标志物对WT患者更好的治疗和更好的整体预后具有重要意义。近年来,ceRNA网络假说引起了研究者的极大兴趣,它将蛋白质编码的mRNA与非编码的RNA联系起来。根据ceRNA假说,circRNA可以作为miRNA海绵来影响下游mRNA的表达[15-16]。越来越多的证据表明,circRNA及其介导的ceRNA调控网络在多种人类癌症的发病和进展中发挥着至关重要的作用[24]。例如,hsa_circ_0014130作为miR-132-3p的海绵,通过调控KCNJ12的表达,促进膀胱癌的肿瘤发生和转移[25];circAGFG1作为miR-195-5p的海绵,通过调节CCNE1的表达,促进三阴性乳腺癌的进展[26]。因此,为了探索circRNA相关的ceRNA网络在WT发生发展过程中的潜在机制,本研究基于临床肿瘤样本的测序数据构建了WT中一个潜在的circRNA-miRNA-mRNA ceRNA调控网络。
3.1 RNA测序揭示WT中circRNA和mRNA的差异表达
到目前为止,还没有研究报道WT肿瘤和正常组织之间DEcircRNA的全面表达。通过下一代测序技术本研究系统性地分析了WT的circRNA表达谱。通过circBase数据库比对,超过一半的circRNA(54.61%)为新发现。根据方法中的筛选标准,共检测到314个DEcircRNA(115个上调,199个下调)。为了验证测序结果,本研究随机选择了12个DEcircRNA在临床肿瘤组织中进行RT-qPCR验证,结果其中7个DEcircRNA呈现出与测序结果一致的表达趋势。另外,本研究也分析了WT肿瘤和配对正常组织之间的mRNA差异表达,共检测到1 612个DEmRNA(1 030个上调,582个下调)。有趣的是大多数DEcircRNA(63.38%)下调,而大多数DEmRNA(63.90%)上调。这些结果暗示一个复杂的ceRNA调控网络。
GO富集和KEGG途径分析表明,DEcircRNA亲本基因富集到多条癌症相关的途径中,参与细胞增殖和细胞周期等恶性生物学相关过程。GO-CC分析提示DEcircRNA亲本基因主要定位于细胞质中。一般认为,大部分外显子来源的circRNA主要定位在细胞质中发挥转录后调控功能。值得一提的是外泌体中也有大量富集。最近的研究揭示了circRNA在外泌体中具有稳定的富集[27]。不仅如此,越来越多的研究表明外泌体来源的circRNA在多种肿瘤中充当预后标志物,并调控肿瘤细胞增殖、侵袭、转移和化疗耐药[28-32]。以上证据支持circRNA通过外泌体运输的方式参与调控WT的肿瘤微环境。GO和KEGG通路分析也用于注释WT中的DEmRNA。结果显示DEmRNA参与调控细胞分裂、细胞周期、细胞增殖以及代谢和经典的P53通路等生物过程,这些信号通路已被证实可以调节细胞的行为和结局。结果表明,DEcircRNA和DEmRNA在多种癌症相关功能通路中起着关键作用,并进一步表明circRNA相关的ceRNA网络可能参与了WT的发生和发展。
3.2 WT中circRNA-miRNA-mRNA调控网络的构建
越来越多的研究发现,circRNA可以作为miRNA海绵间接调控基因表达。因此,为了揭示DEcircRNA在WT中的详细作用和机制,本研究整合了circRNA-miRNA和mRNA-miRNA相互作用对,构建了由62个circRNA、8个miRNA和127个mRNA组成的circRNA-miRNA-mRNA调控网络。为了了解ceRNA网络中的mRNA靶点在WT发生和发展中的潜在途径和生物学功能,本研究进行了GSEA分析。GSEA可以检测基因集合而不是单个基因的富集途径。富集结果主要与细胞周期有关。值得注意的是,这些mRNA靶点还富集到多种免疫细胞活性路径。如上所述,结果表明ceRNA网络中的mRNA靶点可能通过细胞周期和免疫途径调控WT的发生发展。
为了进一步确定关键的调控网络,基于STRING数据库建立了PPI网络,并筛选了10个HUB基因。除了KANK3在WT下调外,其余HUB基因均呈上调。单因素Cox分析发现,ceRNA网络中的靶分子TP53、KANK3、LLGL1和TOP3A是WT患者预后不良的危险因素。进一步表明它们在WT进展中具有重要的生物学作用。
3.3 体外实验验证circRNA调控网络的潜在功能
通过在PubMed上搜索这些基因,本研究发现既往的研究已经报道了他们在恶性肿瘤发生中的可能机制。TP53在多种癌症中均有高表达,并可作为多种癌症的诊断和预后标志物[33]。此外P53也被报道作为circRNA的调控靶点,例如一项最近的研究显示circCNTNAP3的过表达抑制了P53野生型食管鳞状细胞癌 ESCC 细胞的增殖同时增加细胞凋亡,机制上通过海绵miR-513a-5p从而促进P53的表达[34]。相对而言,KANK3的研究较少,KIM等[35]发现KANK3的下调增强了肝细胞癌的细胞迁移和侵袭,生物信息学分析表明KANK3下调与多种癌症的不良预后相关。有趣的是,KANK3已被确定为P53的缺氧诱导的促凋亡靶点[36]。LLGL1也被称为Lgl1、HUGL1或HUGL-1。该基因已被报道通过调节细胞增殖、侵袭、细胞凋亡和化疗药物耐药性等生物学过程在多种恶性肿瘤中发挥致癌作用,比如胰腺导管癌[37]、非小细胞肺癌[38]、神经胶质瘤[39]和食管癌[40]。LLGL1还被报道作为多种癌症的预后标志物[41-44]。TOP3A编码一种DNA拓扑异构酶。有研究报道了该基因在癌症中的作用,例如该基因与P53相互作用并有助于P53介导的肿瘤抑制[45]。另一项研究则发现TOP3A的表达与口腔鳞状细胞癌癌症干细胞和免疫表型之间存在相关性,然而并未显示出该基因在口腔鳞状细胞癌中的预后意义[46]。最后,基于关键HUB基因的重要性,使用RT-qPCR在临床来源的肿瘤组织中验证了他们的差异表达水平。此外,功能分析显示,hsa_circ_0009035的沉默抑制了WT细胞的增殖、侵袭和迁移,提示hsa_circ_0009035在WT细胞恶性进展中的作用。这些发现有力地证明了环状RNA在WT中不可或缺的作用。
本研究确定了WT中的circRNA表达谱并构建了circRNA相关调控网络。该网络可能参与调控WT的发生发展。本研究为阐明WT的发病机制提供了新的见解,并提出了值得进一步研究的潜在调控机制。
猜你喜欢
中国典型病例大全(2022年11期)2022-05-13
中国典型病例大全(2022年7期)2022-04-22
科学导报(2021年29期)2021-06-03
三农资讯半月报(2021年1期)2021-01-27
科海故事博览·下旬刊(2019年6期)2019-04-16
中学生数理化·高一版(2017年2期)2017-04-25
数学学习与研究(2017年3期)2017-03-09
中国医药导报(2016年33期)2017-03-06
中学生物学(2016年10期)2016-11-19
现代养生·下半月(2015年8期)2015-11-16
