
镍催化β-溴-α-氨基酸与溴化氮杂芳烃的交叉偶联反应
2022-10-27 08:04张亚凤黎樱子刘一豪陈妍祺徐振华
化工技术与开发 2022年10期
张亚凤,黎樱子,刘一豪,陈妍祺,徐振华,唐 石
(吉首大学化学化工学院,湖南 吉首 416000)
氨基酸及其衍生物的药理活性作用突出,可用于治疗肝病疾病、脑病、呼吸道疾病等,在癌症治疗方面也取得了新进展,在食品、化妆品和化工等领域也有广泛应用[1-2]。但天然氨基酸自身的功能基团相对缺乏,不能完全满足生物化学等研究领域和生产实际中对蛋白质形态及功能的需求[3-5],因此非天然氨基酸的合成与研究意义重大。
非天然氨基酸含有羰基、不饱和键、无机盐酸根等多种功能基团,可灵活进行很多的修饰反应,其作为关键基序,广泛存在于各种药剂、农药和生物相关的天然产物中[4-7]。因此,探求非天然氨基酸的合成方法具有重要价值,引起了研究者的广泛关注,形成了多种合成方式,其中通过交叉偶联反应构建C-C键的思路新颖,发展空间充足。
Smiles重排是一种分子内亲核芳香取代反应,被证明是合成多种用途化合物的有效途径[8-11]。最新的研究成果认为,Smiles重排反应的反应条件温和,在描述各种芳香族与杂芳香族化合物的合成方面具有优越性[12]。氮杂芳烃是一类重要的化合物,广泛存在于天然产物中,但在氮杂吩噻嗪的合成中,Smiles重排尚未得到全面描述[13]。
基于此背景,本文研究了一种基于Smiles重排的、镍催化的亲电还原交叉偶联制备β-芳基化氨基酸衍生物的方法,有效合成了2-(苄基(喹啉-3-甲基)氨基)3-苯基己酸乙酯和2-(苄基(吡啶-3-甲基)氨基)3-苯基己酸乙酯,合成路线简洁,操作简便。工艺路线见图1。
1 仪器和试剂
仪器:ZF-1型三角紫外分析仪,RE-52AA型旋转蒸发仪,集热式恒温搅拌器,AVANCE 400MHz 型核磁共振波谱仪(NMR)。
试剂:二苄胺、溴乙酸乙酯、苯甲醛、苄胺、正丁醛、正丁基锂、二异丙胺、四溴化碳、三苯基膦、溶剂无水乙醇、乙腈、四氢呋喃(THF)(都要进一步干燥除水);碳酸钾、氯化铵、无水硫酸钠、硼氢化钠、芳基卤、锌粉、4,4′-二甲氧基-2,2′-联吡啶、氯化镁、磷酸钾、三聚氰胺、NiBr2(dme)等(除特别标注外均为分析纯)。
2 实验部分
2.1 中间产物2的制备
在100 mL圆底烧瓶中依次加入20mmol硼氢化钠和6mL无水乙醇,另准备10 mmol苄胺、12 mmol苯甲醛和一定量的无水乙醇,依次加入一个干燥的离心管内,摇匀,静置6min备用。将苄胺与苯甲醛的混合液在搅拌条件下逐滴加入圆底烧瓶中,于80℃反应过夜。经TLC板检验反应完成,用水淬灭反应,用旋转蒸发仪将甲醇旋干,再用乙酸乙酯萃取3次,合并有机相,用无水硫酸镁干燥,过滤,浓缩滤液,得到中间产物2,产率为75%。
2.2 中间产物3的制备
依次将20mmol碳酸钾、10mmol溴乙酸乙酯、15mL乙腈加入100mL圆底烧瓶中,在冰浴条件下,边搅拌边滴加12mmol的中间产物2。观察到溶液浑浊后,加入0.5mL三乙胺,撤去冰浴,于室温下反应24h。将得到的浊液抽滤,保留滤液,分别用5%的氯化钠溶液和蒸馏水各洗涤2遍,保留有机层,加适量无水Na2SO4干燥,过滤、旋干,得到中间产物3,产率为78%。
2.3 中间产物4的制备
依次取14mmol二异丙胺、6mL四氢呋喃加入250mL的Schlenk管中,在-40℃、氮气保护下,注入14mmol正丁基锂,不施加其他外力,使体系在1h内升温到-20℃,再降温到-40℃。将10mmol中间产物3溶于2mL四氢呋喃中,注入Schlenk管中,保温30min,最后注入12mmol正丁醛。将反应移至室温下反应4h后,用饱和NH4Cl水溶液淬灭,乙酸乙酯萃取3次,保留有机层,旋干后得到粗产物,用柱层析法进行分离,得到中间产物4,产率为71%。
2.4 中间产物5的制备
取7mmol四溴化碳、5mmol中间产物4及适量的四氢呋喃于250 mL的Schlenk管中,在-5℃、氮气保护下,将溶有7mmol三苯基膦的2mL四氢呋喃注入Schlenk管中,保温40min后,转移到40℃的油浴锅中继续反应,20h后反应基本结束。经过滤、减压浓缩、柱层析纯化,得到交叉偶联原料5[3-溴-2-(二苄基氨基)己酸乙酯],产率为68%。
2.5 交叉偶联产物的制备
在Schlenk管 中 加 入0.2mmol的 产 物5,与0.3mmol的3-溴喹啉或3-溴吡啶一同作为反应底物,1 mol% NiBr2(dme)为催化剂,加入0.4mmol的Zn粉,0.4mmol的MgCl2,0.024mmol的 配 体4,4′-二 甲氧基-2,2′-联吡啶,0.14mmol三聚氰胺,0.2mmol的碱性添加剂K3PO4,2mL的DMA溶剂,充氮气保护,35℃下反应24h,反应基本完成,经乙酸乙酯萃取及柱层析分离,得到目标产物6。
2-(苄基(喹啉-3-基甲基)氨基)-3-苯己酸乙酯(3a),无色油状液体,产率为65%。1H NMR(400MHz, CDCl3):δ 8.34 (d,J=2.2Hz,1H),8.08(d,J=8.6Hz,1H),7.67(dd,J=8.5,5.7,1.8Hz,2H),7.57~7.43(m,2H),7.41~7.18(m,7H),6.94~6.82(m,4H),4.50~4.31(m,2H),4.04(d,J=14.1Hz,1H),3.83(d,J=13.6Hz,1H),3.59~3.41(m,2H),3.35~3.18(m,2H),1.46(t,J=7.1Hz,3H),1.33~1.25(m,2H),1.11~1.02(m,2H),0.74(t,J=7.3Hz,3H)。13C NMR(126MHz,CDCl3):δ 171.6,151.9,147.3,141.4,138.4,136.0,132.0,129.3,129.1,129.0,128.3,128.0,127.8,127.6,127.2,126.7,126.5,65.6,60.3,54.5,51.7,45.3,36.7,29.7,20.4,14.8,13.9。HRMS m/z (ESI) calcd for C31H35N2O2[M+H]+467.6218,found:467.6217。
2-(苄 基(吡 啶-3-基 甲 基)氨 基)-3-苯 己酸乙酯(3b),无色油状液体,产率为57%。1H NMR(400MHz,CDCl3):δ 8.43(dd,J=4.8,1.7Hz,1H),8.14(d,J=2.2Hz,1H),7.32~7.24(m,3H),7.22~7.16(m,3H), 7.08(dd,J=7.8,4.7Hz,1H),6.98(dt,J=7.7,2.1Hz,1H),6.85(ddd,J=7.1,5.1,1.9Hz,4H),4.36(ddd, J=47.5,10.8,7.2Hz,2H),3.81(t,J=13.8Hz,2H),3.46(d,J=11.4Hz,1H),3.28~3.16(m,3H),1.44(t,J=7.1Hz,3H),1.39~1.30(m,2H),1.09~1.00(m,2H),0.75(t,J=7.3Hz,3H)。13C NMR(126MHz,CDCl3):δ 171.5,150.5,148.4,141.4,138.5,136.8,134.5,129.2,128.9,128.1,128.0,127.1,126.5,123.0,65.6,60.3,54.3,51.5,45.2,36.7,20.4,14.8,13.9。HRMS m/z (ESI) calcd for C27H32N2O2[M+H]+416.5552,found:416.5556。
3 结果与讨论
3.1 条件优化
为了节约成本,提高收率,使反应达到最佳效果,笔者对还原交叉偶联反应的条件进行了优化。首先研究了催化剂对反应的影响。没有催化剂时,反应几乎无法进行,或进行得非常缓慢以至于几乎检测不到产物。对比了NiBr2(dme)和NiCl2(dme),数据显示NiBr2(dme)的产率比NiCl2(dme)高11%,催化效果更好,因此NiBr2(dme)是反应的最佳催化剂。
分析了溶剂的影响。分别将DMA(N,N-二甲基乙酰胺)、DMF(N,N-二甲基甲酰胺)、CH3CN作为溶剂,得到的产物产率分别为65%、62%、59%,由此可知DMA是此步骤的最佳溶剂。
探讨了碱性添加剂对反应的影响。分别选择了3种不同的试剂K3PO4、K2CO3、Et3N,结果显示K3PO4的效果最理想,产率达65%, K2CO3由于碱性较强,产率(61%)不如K3PO4,三乙胺的效果更差,因此K3PO4是较好的碱性添加剂。
对于反应温度,笔者发现35℃油浴下的产率最佳,因此确定该反应的优化条件为:NiBr2(dme)为催化剂,DMA为溶剂,K3PO4为碱性添加剂,4,4′-二甲氧基-2,2′-联吡啶为配体,35℃下油浴反应20h。
3.2 产品的结构表征
图2、图3分别是2-(苄基(喹啉-3-基甲基)氨基)-3-苯己酸乙酯和2-(苄基(吡啶-3-基甲基)氨基)-3-苯己酸乙酯的氢谱图,图4、图5分别是2-(苄基(喹啉-3-基甲基)氨基)-3-苯己酸乙酯和2-(苄基(吡啶-3-基甲基)氨基)-3-苯己酸乙酯的碳谱图。
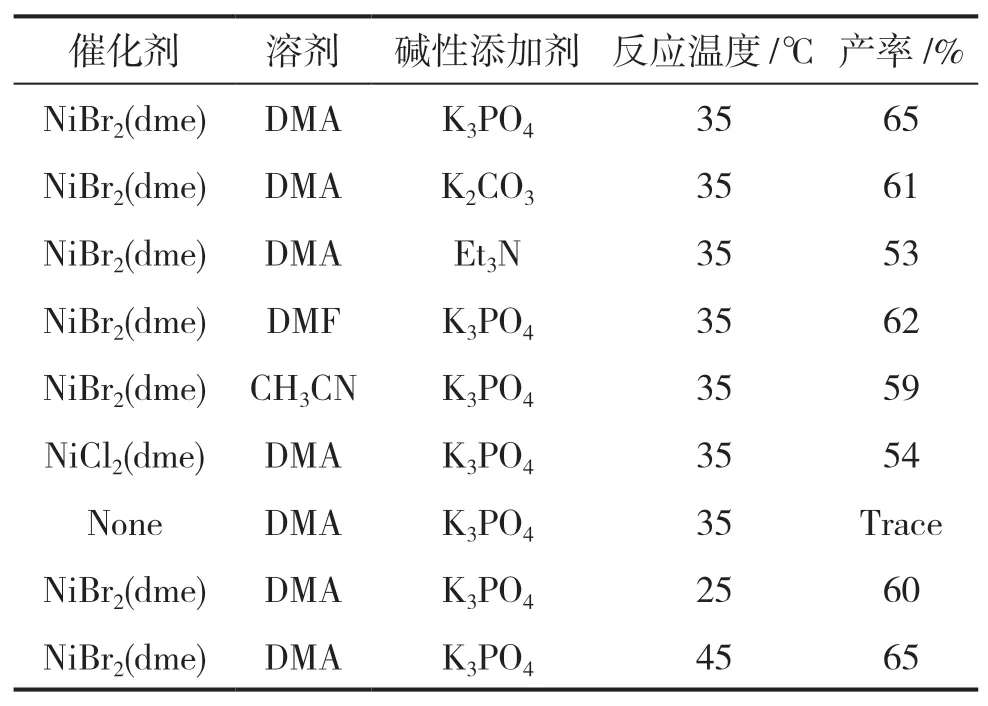
表1 还原交叉偶联反应条件的优化Table 1 Optimization of reduction cross coupling conditions
4 结论
本文研究了基于Smiles重排的、镍催化的亲电还原交叉偶联反应制备β-芳基化氨基酸衍生物的方法,有效合成了2-(苄基(喹啉-3-甲基)氨基)3-苯基己酸乙酯和2-(苄基(吡啶-3-甲基)氨基)3-苯基己酸乙酯,探究了催化剂、溶剂、辅助剂、反应温度等因素的影响,确定了交叉偶联反应的最优条件:以NiBr2(dme)为催化剂,DMA为溶剂,K3PO4为碱性添加剂,4,4′-二甲氧基-2,2′-联吡啶作配体,在35℃下油浴反应20h。此方法的合成路线简洁,操作简便,可为非天然氨基酸衍生物的合成以及亲电交叉偶联反应提供实验数据参考。
猜你喜欢
酿酒科技(2022年8期)2022-08-20
作物学报(2022年7期)2022-05-12
酿酒科技(2021年11期)2021-11-24
药学进展(2021年3期)2021-05-11
药学进展(2021年3期)2021-05-11
井冈山大学学报(自然科学版)(2021年1期)2021-03-05
汕头大学学报(自然科学版)(2020年2期)2020-06-12
酿酒科技(2020年1期)2020-04-30
酿酒科技(2020年1期)2020-04-30
药学研究(2015年11期)2015-12-19
