
万寿竹中典型化合物缓蚀性能理论研究*
2022-12-01 12:48谢鑫钰李晓靖唐情轩何盈春
广州化工 2022年20期
苏 柯,谢鑫钰,李晓靖,丁 壅,唐情轩,何盈春
(阿坝师范学院,四川 阿坝 623000)
钢铁对我国的经济发展有着不可替代的贡献,但是遗憾的是,无论在哪个领域的应用中,腐蚀都无法避免,因此,开发新型的钢铁缓蚀剂有着重要的意义[1]。目前国内外缓蚀剂筛选方法主要还是实验室大规模的探索,此法工作量大,需要大量的化学试剂,不仅存在着资源的浪费,也对环境保护带来了一定的压力。近年来,量子化学计算方法具有效率高,操作简便和节约资源等优点所以备受关注[2]。
相对于常规缓蚀剂来讲,植物缓蚀剂具有来源广泛,绿色环保等突出优点,故研究植物缓蚀剂具有优良的前景[3]。万寿竹Disporumcantoniense(Lour.)Merr.来源广泛,价格低,纵览文献[4]后发现,我们归纳了具有代表性的16个化学物质,这些结构多为多元杂环化合物且具有较大的共轭结构,符合有机缓蚀剂的一般结构要求。
基于此,本文通过对前人总结万寿竹中部分有效成分,从理论上筛选出万寿竹中有效的缓蚀成分,并对其缓蚀机理分析评价[5],为后续万寿竹的开发应用提供一定数据基础。
1 计算方法与模型
利用高斯软件中密度泛函理论,采用 B3LYP 方法,在6-311+g(d,p) 基组水平上,研究万寿竹中16种主要化合物的分子结构及电子结构,计算其前线轨道分布,利用计算结果的量子参数,进一步计算其fukui指数,讨论了其缓蚀活性。
2 结果与讨论
2.1 分子的几何构型分析
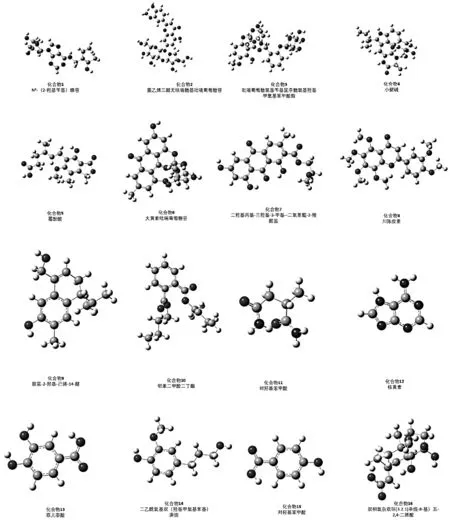
图1 16种万寿竹中典型提取化合物的结构
优化后我们发现,万寿竹中主要结构可以分为三类:第一类为具有三个及三个以上环平面的大共轭结构,包括化合物2、4、5、6、7、8、9和12;第二类化合物虽然具有较大的平面的共轭体系,但是边缘具有簇状及笼状结构,包括化合物1、3和16;第三类化合物是单环结构或未成环结构,包括化合物10、11、13、14和15。总的来说,三种化合物都具有p-π共轭或π-π共轭现象具有p-π共轭或π-π共轭现象,且第一类大于第二类大于第三类,而这种共轭结构为化合物与金属之间发生的电子转移,提供了可能,从而使化合物吸附在缺电子金属上起到缓蚀作用。以此理论为基础,第三类化合物共轭体系还不够高,故第一和第二类化合物更符合缓蚀剂的一般结构要求。
2.2 缓蚀活性分析
2.2.1 量化基本参数分析
在上述分子的优化结构上,进一步计算了所有分子的总能量(tatol energy)、偶极矩(dipole moment)、最高占据轨道能量(EHOMO)、最低空轨道能量(ELUMO)以及前线轨道能量的差(ΔE),所有数据如表1所示。
根据分子前线轨道理论,最低未占据轨道能量(LUMO)与最高占据轨道能量(HOMO)的差称为分子能量间隙,由于多数金属缓蚀都与吸附相关,而分子能隙在化合物的吸附行为中起着重要作用。HOMO的能量显示出给予电子的能力,而LUMO的能源显示出接受电子的能力。金属很容易接受来自电子供体(例如抑制剂分子)的电子进入LUMO。相反,金属可以通过反键将其HOMO电子贡献到抑制剂分子的适当空位轨道(LUMO)中。抑制剂分子和金属轨道之间的供体-受体关系是抑制剂分子在金属表面吸附背后的主要分子和电子解释。当HOMO和LUMO与软钢表面铁原子的导带和价带相互作用时,吸附行为完成。由表1分子能隙ΔE数据可得化合物6<3<2<16<1<4<7<8<5<9<15<12<14<11<13<10。数据表明,化合物6能隙低于1 eV,说明其分子稳定性较差。而其他化合物能隙高于1 eV,所以容易与金属发生吸附作用。
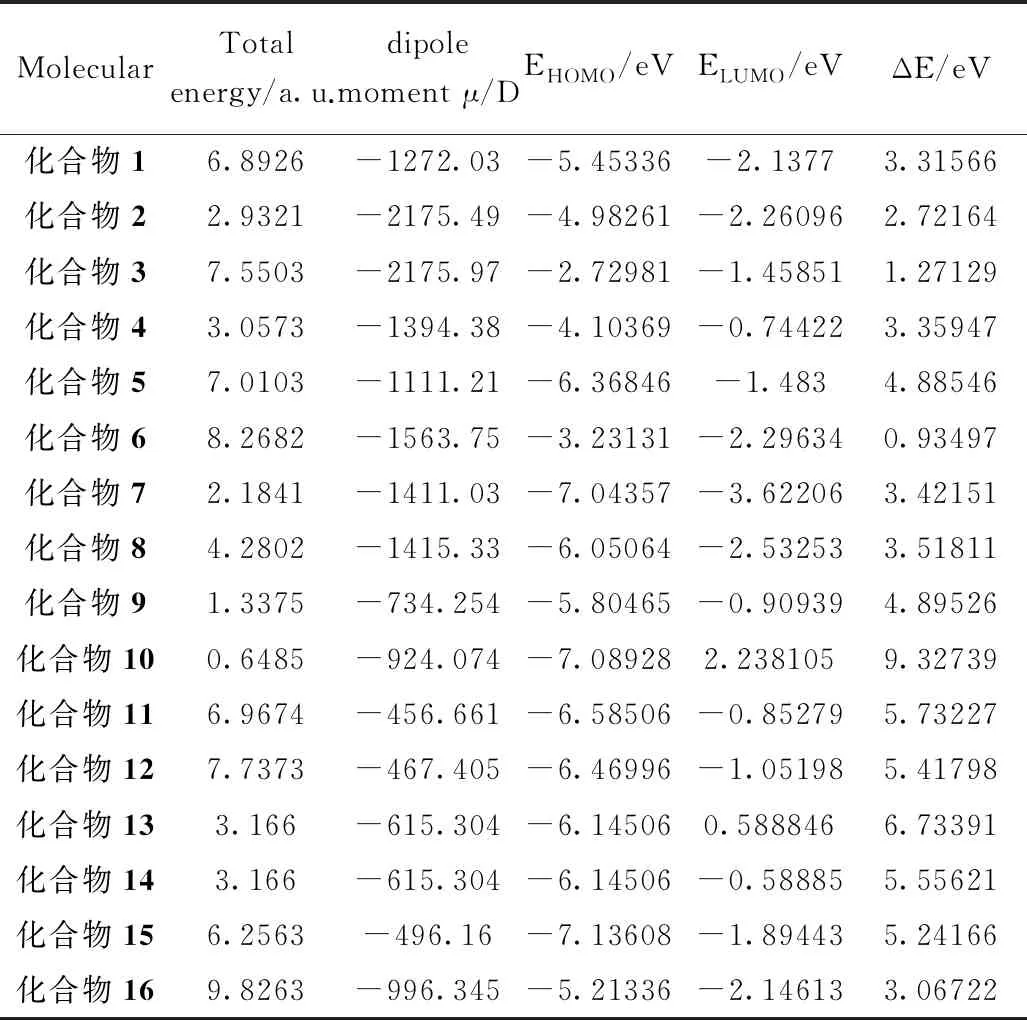
表1 分子的总能量、偶极距、最高占据轨道能量、最低空轨道能量和能隙
计算过程中,通常认为在物理吸附中,分子的极性越大,缓蚀剂的缓蚀效应越强。在计算过程中我们以偶极矩的数值来理解化合物分子的极性,因此,根据分子极性来推测万寿竹中典型化学物质金属表面发生的物理吸附的难易程度为:化合物16>6>12>3>5>11>1>15>8>13>14>4>2>7>9>10。从排序和数据可以看出,相对来说第二类化合物即化合物1,3和16极性相对较大,容易发生物理吸附,化合物10结构较为对称,极性较小,不容易发生物理吸附作用。
为了深入的了解万寿竹中的典型化学物质的缓蚀机制,我们利用Hartree-Fock理论[5],计算了万寿竹中的典型化学物质与金属Fe的电子转移数和亲电子能力。相关电子转移数可以和衡量一个分子的亲电子能力大小的亲电指数ω可以通过如下公式计算:
ΔN=(χFe-χinh)/2(ηFe+ηinh)
ω=μ2/2η
这里的离子化电位(I)近似等于-EHOMO,电子亲合势(A)近似等于-ELUMO。而全域硬度(η)和电负性(χ),则被近似定义为η=(I-A)/2,χ=-μ=(I+A)/2。
从表2我们可以发现,万寿竹中典型化合物的亲电指数都高于2 ev,说明所有化合物都具有较高的亲电性,易与金属Fe发生亲电反应。结合文献调研分析综合来看,第一类化合物中化合物5,7和8除具有大的电负性和亲电指数外,还具有较高的全域硬度和电子转移数,而第二类化合物中电负性和亲电指数相对较低,这说明可能是容易发生亲核反应的原因。文献报道,当金属和缓蚀剂分子相互靠近时,电子将从低电负性位置转移到高电负性位置,直到平衡。当ΔN值为正值时,电子从缓蚀剂分子转移到低碳钢表面的铁原子,所以,除了化合物10和13,其他分具有良好的给电子能力,并与接受电子的Fe离子表面通过配位键的形式发生作用。
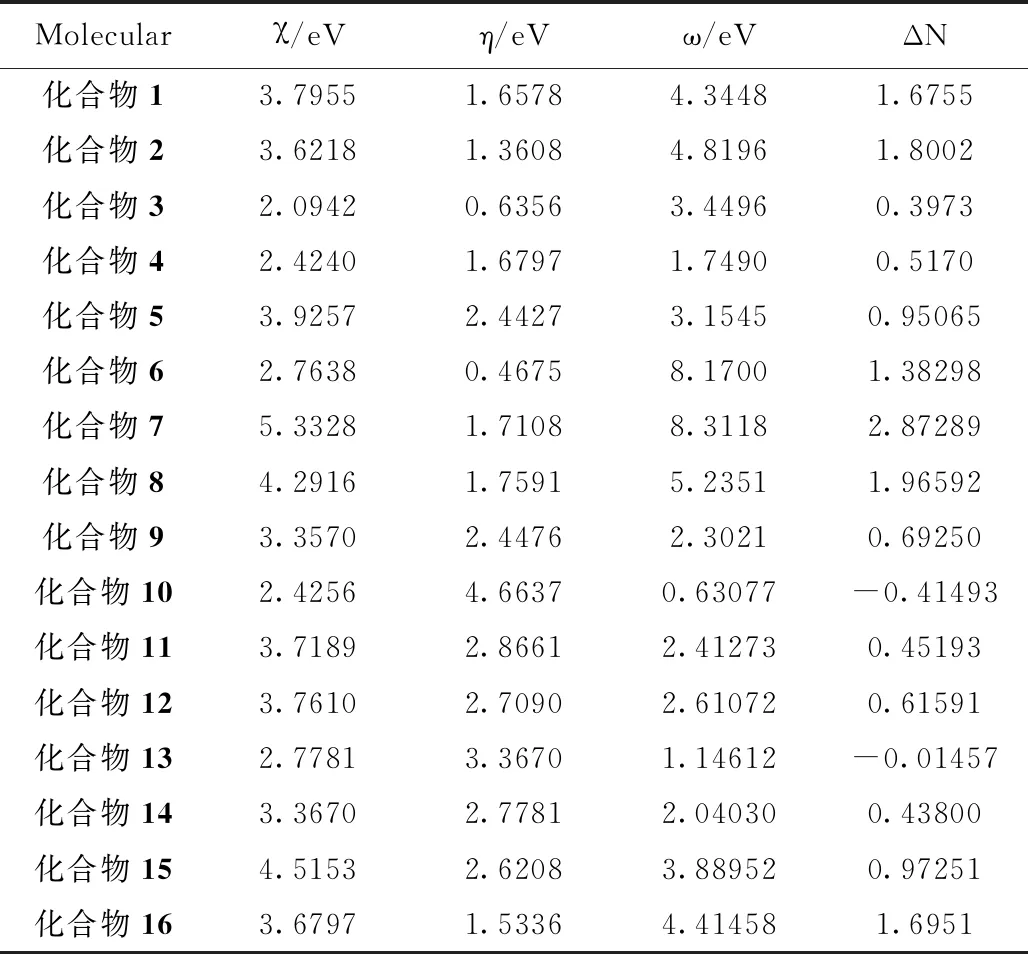
表2 电负性χ和全域硬度η、亲电指数ω和电子转移数ΔN
2.2.2 分子静电势
分子静电势可以直观反映化合物的反应活性位点,静电势越正,亲电活性越大;静电势越负,意味着亲核反应活性越大。
根据图2万寿竹中典型提取化合物的负静电势不难发现,化合物中电子基本集中在含有O原子官能团附近,说明结构中的氧原子应该是吸附在铁上面的反应活性位点。其中共平面性强,偶极矩大,稳定性高的第一类物质中的化合物5,7和8静电势分布类似,分散在化合物边缘的氧原子附近,说明在吸附过程的时候,化合物5,7和8容易在分子内羰基附近,发生亲电反应,这与它具有大的电负性和亲电指数和较高的全域硬度和电子转移数数据相吻合。

图2 16种万寿竹中典型提取化合物的表面负静电势
3 结 论
本实验研究了万寿竹中16种主要化合物的分子结构及电子结构讨论了其缓蚀活性。从优化机构来看,化合物10、11、13、14和15中共轭体系较小,理论上缓蚀性能不高,从分子能隙数据来看,除化合物6以外都容易发生吸附作用,而从偶极矩数值来看,化合物1,3和16极性相对较大,容易发生物理吸附,而根据Fock方程计算结果,化合物5,7和8具有大的电负性和亲电指数外,还具有较高的全域硬度和电子转移数,容易发生亲电反应,通过静电势上的数据我们发现,化合物5,7和8的反应活性位点还是集中在羰基氧原子附近,与之前的推测一致。本项目为缓蚀剂的开发提供新思路,为万寿竹作为缓蚀剂的开发和应用提供一定的参考依据。
猜你喜欢
大电机技术(2022年3期)2022-08-06
都市人(2022年3期)2022-04-27
国际太空(2021年8期)2021-11-05
炼油与化工(2021年5期)2021-10-19
安全、健康和环境(2020年10期)2020-11-18
当代化工(2019年1期)2019-12-12
环球时报(2019-12-05)2019-12-05
中学化学(2017年6期)2017-10-16
中学化学(2017年6期)2017-10-16
中学化学(2017年2期)2017-04-01
