
复旦大学附属儿科医院高通量测序数据一体化全流程闭环分析系统及临床应用案例分析
2022-12-15 02:00董欣然王慧君吴冰冰王雅琼周文浩卢宇蓝
中国循证儿科杂志 2022年3期
陈 宾 董欣然 王慧君 吴冰冰 杨 琳 王 潇 王雅琼 倪 琦 李 川 周文浩 卢宇蓝
高通量测序技术或称下一代测序技术(NGS),自2010年起应用于遗传病的临床遗传检测[1-3]。根据构建测序文库的方法不同,检测基因组DNA的NGS分为外显子组捕获测序(ES)和全基因组测序(WGS)。ES富集外显子区进行测序,包括全外显子组测序(WES)、临床外显子组测序(CES)和基因包测序(panel)。WGS则直接测序整个基因组DNA。ES主要检出单核苷酸变异(SNV)和小插入/缺失(Indel),也可分析拷贝数变异(CNV)[4,5]和杂合性缺失(LOH)[6]等特殊类型的变异, 但检测范围仅局限在捕获区域附近。WGS除了能检测ES所覆盖的变异类型之外,还能检测到更复杂的结构变异且检测范围覆盖整个基因组。
本研究团队于2015至2018年建立了ES的临床遗传病诊断分析的高通量测序数据分析流程(简称复旦流程1.0和2.0)[7,8],通过变异注释和变异筛选辅助临床快速锁定潜在致病位点;为改善WGS的成本和效率问题,本研究团队也设计了针对危重症的快速全基因组流程(rWGS)和经济版家系全基因组测序方案(OTGS)[9,10]。由于ES和WGS在性价比和变异检测范围等方面各有优势,因此建立一套能够支持多种测序方案的分析流程是提高遗传检测效率的关键。本研究建立了一体化数据复旦流程3.0,并通过一组临床测序病例的实际应用,测试其在变异检测范围、变异优选效率、数据分析效率等方面的效果。
1 一体化分析流程建立
整体分析流程包括7个主要功能模块:①病史处理,②结构化遗传表型,③测序实验,④变异检测,⑤变异解读,⑥质控复核,⑦基因型表型联合分析。图1显示流程的整体架构和模块间互通概况。
1.1 临床病史传递与结构化处理 包括病史处理和结构化遗传表型2个模块(图2)。病史处理模块对接医院信息系统,自动提取检测申请信息并加密。其中,与检测申请相关的信息对接样本库,用于自动生成报告草稿;检测类型、样本性别和亲属标识等信息用于数据质控复核。现病史等文本数据对接结构化遗传表型模块,从Auto-Neo-HPO系统[11]自动提取HPO词条,传递至图1中基因型表型联合分析模块。
1.2 样本处理与测序实验 DNA的抽提使用血液基因组DNA提取试剂盒(TIANGEN公司),DNA浓度的检测使用 NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA) 和Qubit Assay (Thermo Fisher Scientific, USA)。图3显示测序实验模块的功能细节,WGS、WES和CES检测对应的DNA文库分别为TruSeq Nano DNA Library Prep Kit (Illumina, cat no. 20015965)、IDTxGenomes2和Berry’s NanoWES Human Exome。采用Illumina NovaSeq 6000平台测序,下机数据为FastQ文件。

图1 复旦流程3.0整体设计和模块间交互关系示意图
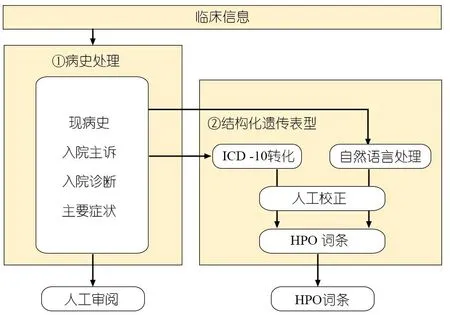
图2 临床病史传递与结构化处理示意图

图3 测序实验模块的细节设计和信息传递
1.3 根据文库应用变异检测 图4显示,变异检测模块功能细节和变异检测间关联。变异检测模块基于现有生物信息学算法工具,辅以信息传递、资源调配、进度控制等功能。WGS变异检测于Dynamic Read Analysis for GENomics Bio-IT(DRAGEN)平台完成(搭载CNVNator和MANTA算法),产出变异为SNV(含线粒体)、Indel、CNV和结构变异(SV);ES的变异检测由Sentieon系统分析SNV(可含线粒体),Indel和CANOES流程分析CNV[5,12]。此外,可基于SNV检测LOH变异,基于CNV中间文件补充SMN1和SMN2分析结果[13]。
1.4 数据质控与变异复核验证 数据质控模块在测序数据下机、变异检测结束、变异解读分析后都会介入。从实践

图4 变异检测模块功能细节
数据质控以BAM文件内容为主,包括匹配短序列数量、捕获区域覆盖百分比、平均覆盖乘数和>20×的覆盖区域百分比,数据质控的阈值会综合考虑所用建库方案、上样量、测序实验流程等。变异检测的结果也可用于数据质控。基于CNV分析,可对样本的性染色体拷贝数和SRY基因进行复核;基于SNV检测,可评估样本污染情况及样本间亲缘关系[14]。
变异复核验证在SNV、CNV、SV和LOH间展开,如图4中虚线箭头所示。在SNV变异的核型结论和CNV间进行复核,标注结论不一致的变异(如杂合缺失CNV中覆盖的杂合SNV);复核CNV与涉及拷贝数变化的SV、子代染色体末端的CNV与亲代的平衡易位SV,标注结论一致的区域。复核LOH区域和CNV杂合缺失区域,去除带有LOH信号特征但覆盖CNV杂合缺失的区域。

图5 质量复核模块示意图
1.5 变异注释与筛选流程 与ES相比,WGS数据检测到的变异种类显著增多。根据变异类型特点,按SNV/Indel、CNV、SV、线粒体和LOH 5类分别进行变异注释与筛选流程,并在涉及复杂变异组合的场景将其他类型的变异统一汇总到SNV/Indel进行分析。适用于非编码区域变异的注释内容和筛选逻辑,并按照不同建库方案建立子人群数据库,实现不同测序方案的统一管理。具体的注释与筛选流程如下。
1.5.1 SNV/Indel分析流程 SNV/Indel分析流程包括变异质控、有效分析区域控制、人群频率分析、变异危害性分析和遗传模式分析5个步骤。
对于SNV/Indel变异质控问题,基本沿用复旦流程2.0版本[8]并作如下改进:①结合分析SNV和Indel以排除Indel假阳性;②左对齐并校正Indel的5’端接头序列;③更新dbSNP数据库至150版本。
由于WGS的有效测序区域为整个基因组(相对于外显子组增加约30倍),建立了“有效分析区域”来维持分析效率并保留流程对非编码区变异的分析能力。该区域定义为如下并集:所有RefSeq转录本中exon区域双端外延15 bp后的区域、CADD预测分值>20的SNV/Indel、SPIDEX预测Z值绝对值大于1.96的SNV、HGMD数据库标注为DM/DM?、/DP/DFP/R的区域,以及ClinVar数据库有记录且不为“Benign”的区域。位于有效分析区域外的SNV/Indel位点不再进入后续诊断分析流程。
公共数据库及本地人群频率注释包括有效分析区域内的所有位点。不同的建库流程有独立的本地人群子库以排除系统误差;分层的频率筛选阈值基于子人群库的样本量级:子库样本<1 000例时,罕见变异和多态性位点的阈值分别为7.5%和15%,子库样本>1 000例时,阈值分别降至2%和5%。
变异危害性注释和筛选沿用复旦流程2.0版本的变异筛选策略[8],但改进了变异到转录本的映射选择。对于变异到转录本的多重对应问题,对变异映射到的基因、转录本类型、变异HGVS命名分别进行了排序,优先级分别为“已报道致病变异的基因、致病功能明确的基因、基因上变异造成的危害性严重程度”,“NM、XM、NR的转录本”,“已报道变异所用转录本、同基因报道常用转录本、受变异影响危害最大的转录本”。最终,将排第1的转录本所对应的变异危害性类型视为该变异的最终危害性。经此步骤筛选后进行全库的家系关系分析,识别可能的重复送检、家系标注错误等情况,并反馈至图1中质控复核模块处理。
遗传模式分析中纳入了更为复杂的变异类型,当某个隐性遗传的致病基因上检测到致病/疑似致病的SNV/Indel变异时,需标注该基因是否也受到CNV、SV或LOH影响。此外,所涉及的致病基因遗传模式与OMIM数据库同步。
1.5.2 CNV分析流程 CNV分析沿用了前期基于ES数据的分析流程PICNIC[12,15]。对于WGS来源的CNV,保留其原始信号,但在后续分析时降至外显子层面以便与ES数据整合。升级了本地CNV频率库,从拷贝数(0、1、3拷贝)和检测方案(WGS、WES、CES)两方面组合共计9个类别,记录每个类别中每个外显子的CNV检出事件,以评估CNV的人群频率。检出的CNV与SNV/Indel结果相互整合,以提示在常染色体隐性致病基因中可能发生的SNV+CNV复合杂合事件或是解释看似单亲来源的子代纯合变异。
1.5.3 SV分析流程 SV分析一直是WGS分析的难点。从临床解读的角度出发,主要关注结构变异中的CNV与易位变异。流程使用的检测算法为CNVnator[4]和MANTA[16],分别从覆盖深度和读数易位/断裂两方面进行评估,结合两者结论进行复验以提高检测精度。对于不引起CNV的平衡易位断点,则去除内部频率>0.2%的断点,用于标记受累基因并加入SNV/Indel分析流程。
1.5.4 LOH分析流程 单亲二倍体(UPD)涉及的区段可表现为大量连续纯合SNV,即LOH。WGS能获得丰富的SNV信息来检测杂合性丢失,ES在捕获密度较高的区域也能产出足够的信号。在BCFtools/RoH软件[17]的基础上建立了拼接纯合区段和注释印记基因的流程,并通过可视化变异BAF(B-allele frequency)呈现LOH结果,辅助识别致病或提示UPD的LOH[18,19]。
由于杂合性拷贝数缺失(Het-Del)也会导致大量连续纯合变异,因而可能导致LOH分析的假阳性,但反之也可运用LOH信号进一步复验Het-Del。结合CNV与LOH结果,既提升CNV中Het-Del的可靠性,又过滤由于Het-Del导致的LOH假阳性。
1.5.5 线粒体变异分析流程 WGS和有对应探针的ES能够检测到线粒体基因组中的致病变异。相对于核基因组变异,线粒体变异的检测分析具有特殊性。一方面,线粒体基因组长度较短(仅16 kb),因此其变异数量相对较少,分析的难度在于注释及致病性评估;另一方面,线粒体变异不具有核基因组变异的核型特征,而是呈现为异质性(Heteroplasmy)状态。
在处理线粒体变异时,仅考虑线粒体基因组中的SNV/Indel,用VEP软件注释线粒体基因、变异影响类型、gnomAD的WGS子库中的检出频率、MitImpact数据库标记、氨基酸改变、危害性预测、ClinVar既往报道、疾病表型和变异致病性分类(Mitomap数据库)等。鉴于线粒体变异检出量较少,不再筛选。
1.5.6 其他特殊检测与变异类型合并 NGS在处理高同源序列时会产生错配(mis-mapping)的误差,脊髓性肌萎缩症(SMA)是一个代表性例子。针对SMA的检测问题,融合了针对性算法[13]来校正SMN1和SMN2的拷贝数并结合SNV/Indel进一步分析。
图6显示,变异解读模块功能细节,融入了多种变异类型,在分析隐性遗传模式的致病基因时,采用了2种方式综合考虑多种突变类型。第一,将其他类型的变异汇入SNV/Indel变异标注上,辅助识别变异类型间形成的复合杂合模式;第二,平行展示多种类型变异结果,供灵活查阅。
1.6 表型分析与候选基因排序 图7显示,基因型表型联合分析模块的设计原理和输入输出。该模块应用了基于临床表型预测潜在致病基因的新一代贝叶斯模型[20],结合了该算法的整体表型符合度和前述变异分析流程的筛选结果,可高效锁定致病基因。
2 代表性病例纳入与分析指标
代表性病例的选择基于变异类型和诊断难度,由复旦大学附属儿科医院(我院)分子医学中心提供,分析过程中的个人信息均已去隐私处理。对代表性病例在复旦流程3.0处理过程中的总结展示包括:各模块的输入和输出总结,最终形成报告草稿时的变异结论及临床表型,以及对新的整合性分析流程结果进行评估。

图6 变异解读模块功能细节
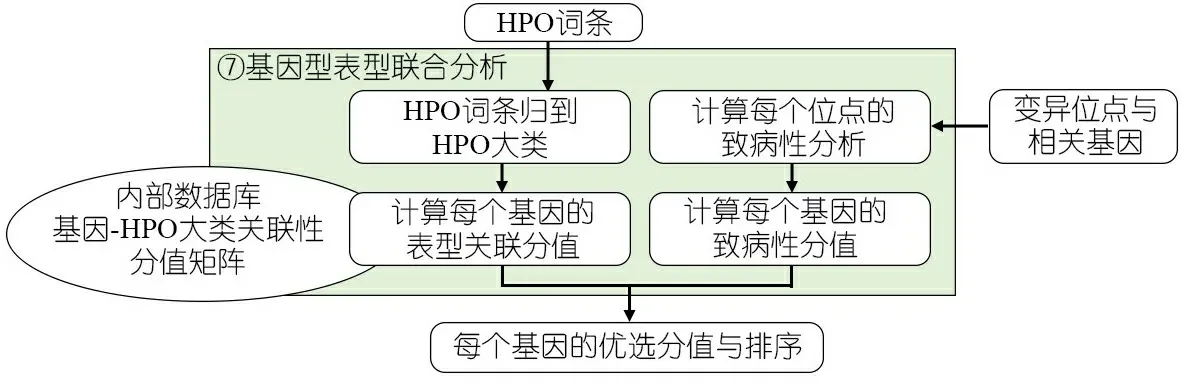
图7 基因型表型联合分析模块的设计原理
2.1 代表性病例
例1:男,初诊3月10 d,因“生后发现运动受限”于我院就诊。3月龄时发现四肢活动少,不能完全抬离床面,查体发现四肢肌力、肌张力低下,下肢屈曲姿势,全身触痛明显,被动上抬肢体不能;X线片提示四肢长骨多发骨膜增生反应,B超提示左肾积水。7月龄时仍不能抬头、翻身,下肢畸形较前加重,双膝、双踝、双肘关节活动受限。
例2:男,初诊5月龄,因“发育迟缓”于我院就诊。其母亲孕期无异常,患儿无出生窒息抢救史。初诊时抬头不稳,查体提示四肢肌张力偏高。头颅MR提示豆状核、丘脑、脑干对称性改变;血质谱检测乳酸偏高,尿质谱未提示异常,临床考虑代谢性疾病可能。
例3:女,初诊3月29 d,因“肌无力1月余”于我院就诊。初诊时四肢无力表现,查体四肢肌力、肌张力低,肌电图提示运动神经病变,考虑SMA。
2.2 代表性病例在复旦流程3.0中的应用 表1显示,例1~3在处理过程中的各阶段信息总结展示,各模块的输入和输出具体数值,报告草稿变异结论和所匹配的临床表型。
例1进行了OTGS,自动化处理提取的HPO词条如下,HP:0000126:Hydronephrosis (肾积水), HP:0001643:Patent ductus arteriosus (动脉导管未闭), HP:0001410:Decreased liver function(肝功能异常),传递给图1中基因型表型联合分析模块。
例1的数据重分析从图1中测序实验模块产出的FastQ文件开始,其质控结果见表1。FastQ数据提交至图1中变异检测模块的WGS分析流程。其中的BAM文件对应质控包括:有效数据量、常染色体及线粒体平均覆盖深度、基因组中10×以上及20×以上覆盖度比例和测序读段与参考基因组的对比率(表1)。OTGS方案的预期测序深度为:先证者40倍、父母各10倍,相关质控数据指标符合预期。
OTGS的变异检测由DRAGEN平台执行,分别产出SNV/Indel、CNV和SV的结果(图4)。变异检测包括SNV/Indel、CNV、SV断点、线粒体变异、LOH、SMN1和SMN2基因拷贝数(表1)。其中,3份样本的性染色体拷贝数结果均符合样本送样标记,先证者与父母之间的亲缘关系符合预期。所测得变异按类型呈递给变异解读模块进行注释和筛选,并汇集到变异相关的致病基因。其处理得到的变异相关基因、所涉SNV/Indel和CNV数量见表1。变异相关基因传递至图1中基因型表型联合分析模块,综合评分Consistency_Score达推荐值>0.3的基因仅有ANTXR2,相关变异为母源遗传SNV:NM_058172: c.1294C>T(p.R432X)、父源遗传SV:4q21.22 del 13 kb,以复合杂合模式对ANTXR2产生影响。ANTXR2基因是玻璃样纤维瘤病综合征(HFS)(MIM:228600)的致病基因;该病呈常染色体隐性遗传,病理变化为细胞外基质在皮下等组织进行性沉积导致,临床表现为丘疹或结节、牙龈肥大、反复感染、难治性腹泻、进行性关节挛缩和骨质疏松等,符合先证者临床表现。

例1,从数据量庞大的家系WGS数据出发,通过把SV和SNV/Indel变异汇聚到基因层面,结合临床病史准确识别出致病变异,综合优选将候选基因排至第1。既解决了常规单独分析SNV/Indel问题可能导致的漏诊,也展现了综合临床表型优选致病基因的效率优势。
例2行先证者单人WGS,自动化提取到HPO词条如下,HP:0001298:Encephalopathy(脑病), HP:0001263:Global developmental delay(全面发育迟缓), HP:0001250:Seizures(惊厥), HP:0001249:Intellectual disability(智力发育迟缓), HP:0006872:Cerebral hypoplasia(大脑发育不全), HP:0000750:Delayed speech and language development(语言发育迟缓), HP:0003198:Myopathy(肌病),传递给图1中基因型表型联合分析模块。
例2的数据重分析从FastQ文件开始,质控数据类型同例1。单人WGS的预期测序深度为40倍,表1可见相关数据指标符合预期。单人WGS的变异检测由DRAGEN平台执行,变异检测类型、质控复核及注释同例1,具体数值见表1。变异相关基因传递至图1中基因型表型联合分析模块,没有综合评分达推荐值>0.3的基因。综合排名前5的基因分别为FLG、SCTLA4、KLHL24、CHEK2和BTG4。其中,FLG基因内部无表型案例较多,SCTLA4基因变异质量欠佳,KLHL24基因与本例先证者表型无关,CHEK2基因为癌症相关综合征基因,BTG4基因为卵母细胞成熟疾病相关基因,结合例2临床分析,上述基因均不能构成诊断。然而在先证者线粒体变异分析中检出了MT-ND6基因上已知致病变异m.14459G>A,变异覆盖深度4 736倍,变异异质性>99.5%,可致Leigh综合征(MIM:256000),符合先证者代谢性脑病表现。
例2,从数据量庞大的单人WGS数据出发,排除了可能的致病性SNV/Indel、CNV和SV变异,并运用WGS的覆盖优势,检测到线粒体高异质性的已知致病变异,展现了覆盖广泛的变异类型优势。
例3进行了单人临床ES,自动化从病史提取到HPO词条:HP:0001324:Muscle weakness (肌无力), HP:0003202:Amyotrophy (肌萎缩), HP:0003202:Skeletal muscle atrophy (骨骼肌萎缩)传递给图1中基因型表型联合分析模块。
例3的数据重分析亦从FastQ文件开始,在图1中变异检测模块进行ES流程分析,产出BAM文件对应质控数据的覆盖范围与WGS不同,具体结果见表1。例3的预期测序深度为150倍,相关数据指标符合预期。变异检测结果见表1,其中SMN1拷贝数估计值为0.775, 提示SMN1杂合缺失可能。
经过图1中变异解读模块分析后,汇集到变异相关的致病基因。经图1中基因型表型联合分析模块,有4个基因(SCN8A、SMN1、NSD1和DYRK1A)综合评分>0.3。其中,SCN8A基因变异表型与先证者不完全匹配,被列为“致病性不明变异”;SMN1基因检出纯合的致病变异NM_000344: c.863G>T(p.R288M),结合CNV检测得出的SMN1基因单拷贝缺失结论和先证者的SMA,被评为“致病性变异”。NSD1和DYRK1A基因变异均在无表型父母样本中检出,不能构成遗传诊断。检出致病变异的基因SMN1是SMA 1~4型(MIM:253300, MIM:253550, MIM:253400, MIM:271150)的致病基因;该病是由脊髓前角运动神经元退行性变而导致的进行性、对称性肌无力和肌萎缩的一类常染色体隐性遗传性疾病,主要表现为肢体近端和躯干肌肉无力和萎缩,符合先证者的临床表现。
例3,依托针对性的SMN1、SMN2基因拷贝数评估能力,SMN1单缺失合并单拷贝致病变异的罕见病例获得了精确诊断。而目前用于诊断SMA的金标准方法多重连接探针扩增技术(MLPA)仅能检出先证者的SMN1单缺失而导致了假阴性。例3展现了对特殊变异的处理能力和结合多种变异类型结果的分析优势。
3 讨论
3.1 高通量测序技术发展方向 高通量测序技术在临床遗传病诊断和研究的发展,主要体现在两个方面。①测序技术,主要的创新为提升文库建库效率、通过标记文库以实现联合分析或精细分析[21-23],以及更新测序技术平台。②数据分析,主要包括从原始测序数据到变异分析的算法更新和遗传变异解读的更新。算法更新主要聚焦分析的速度、精确度和特殊变异类型算法,例如提升SNV/Indel检测效率的Sentieon方案和DRAGEN方案、基于WGS数据分析结构变异的ClinSV流程、基于ES分析CNV的CANOES等;遗传变异解读一方面受益于基因型表型研究的丰富积累(如以OMIM数据库为代表的遗传性疾病表型-基因关联数据库、以GeneCards为代表的整合性数据库、以HGMD/ClinVar数据库为代表的变异解读结论库),另一方面得益于机器学习模型在复杂特征空间下所带来的各种变异危害性评分算法(如CADD[24]、REVEL[25]等)。但是在具体临床应用中,如何协调各系统的优势、调试最佳性能参数、制定最符合应用需求的解读方案,一直是各类技术系统转化到临床应用的“最后一公里问题”。针对这一问题,各医疗和检测机构提出了迭代的解决方案。例如2013年首次提供外显子测序临床应用范式的贝勒医学院[26]、NextCode平台、助力英国十万人基因组计划的Congenica等。这些方案从不同程度缓解了原始变异位点到临床解读的障碍,促进了疾病遗传谱系研究,并帮助发现新的致病基因。
3.2 复旦流程3.0优势 复旦流程3.0是在复旦流程1.0[7]和2.0基础上发展进步中的产物[8]。完成了从“如何实现”、“如何更快”,到现阶段“如何更广”的转变。复旦流程3.0能对接来自各类建库的原始数据,并相应适配分析多种变异类型,再汇聚到病例层面进行辅助诊断。通过3个典型病例的重测分析应用,展示了对于家系WGS、单人WGS和ES测序数据的适配效果,以及对于SNV/Indel、CNV、SV、线粒体变异、杂合性丢失、部分假基因等多种变异类型或特殊情况的处理能力。同时,复旦流程3.0版也从临床应用的角度出发,应用基于复合表型的致病基因排序算法进一步提升解读效率、增加对样本质控的数据分析和对接自动报告系统以降低人为失误,展示了临床遗传检测作为整体打包的可能性,为更多辅助工具和智能算法的应用准备了平台基础。
3.3 复旦流程3.0后续改进方向 作为合并复杂数据来源并覆盖多种变异类型的初步尝试,本流程尚存诸多待优化之处。
首先,SV的亚类变异检测和分析仍有待突破。虽然SV往往作为单独的变异类型被提及,但其包含了多种各具复杂遗传学特征的变异种类,例如缺失、重复、倒位、水平转移和序列串联等,这些变异的成因、遗传学特征、被NGS检测的信号原理以及致病性分析评判标准都各有特点。类似现阶段其他SV分析流程(如ClinSV[27]),复旦流程3.0主要分析SV中的缺失/重复,辅以染色体易位断点进行遗传模式补充,其他类型的SV检测仍有待补充。

最后,在遗传变异的临床解读方面,更便捷的变异注释和分级体系仍有待开发。目前已有较多的变异解读和诊断指南,包括一般性的变异类型解读指南(如ACMG的外显子指南[28]、CNV指南[29]、UPD指南[30])和针对疾病大类的诊断指南(如免疫学会的遗传诊断指南[31])。由于本研究试图建立的流程涵盖了多种变异类型且没有界定遗传性疾病的种类(如只针对遗传性耳聋[32]),因此在变异解读方面仅筛除了良性/可能良性变异和丰富变异位点的注释信息,仍有大量的临床意义未明变异需人工分级。而在结合变异类型分析时,本研究选择以数据量大、已知致病性变异比重高的SNV/Indel变异作为其他变异合并标记的主体,其他变异类型则仅注释到基因层面。
4 小结
本研究从分析多种变异类型、标准化与结构化遗传表型以及以临床分子诊断的变异分析这3方面,聚焦目前广泛应用的ES和WGS测序技术的数据分析问题,建立了从检测申请到报告反馈的复旦流程3.0系统。同时,通过典型诊断病例的重分析,展示了在数据兼容性、流程整合的交互性和为遗传咨询提供信息的有效性方面的具体效果。复旦流程3.0的建立可为未来较长时间内的ES和WGS测序在临床遗传检测领域并行应用提供良好的平台支撑。
猜你喜欢
军事文摘(2022年16期)2022-08-24
中国现代医生(2022年21期)2022-08-22
今日农业(2021年11期)2021-08-13
天津医科大学学报(2021年1期)2021-01-26
中国生殖健康(2020年4期)2020-12-09
医药前沿(2020年20期)2020-11-10
中西医结合肝病杂志(2020年2期)2020-10-27
支部建设(2020年15期)2020-07-08
三农资讯半月报(2020年2期)2020-03-09
百科知识(2015年18期)2015-09-10
