
MnaCe1-aOx纳米棒的制备及其催化氧化CO性能
2022-12-15 04:46朱君江夏明桂
化工环保 2022年6期
吴 婷,高 唯,程 锴,朱君江,2,夏明桂
(1. 武汉纺织大学 化学与化工学院,湖北 武汉 430200;2. 湖北省生物质纤维与生态染整重点实验室,湖北 武汉 430200)
随着城市化进程的加快和工业的迅速发展,大气污染问题日益严重,对生态环境和人体健康造成了极大的威胁[1-3]。汽车尾气是大气污染的主要来源之一。目前用于汽车尾气处理的贵金属催化剂存在成本高和高温易失活等问题,因此开发相对廉价且高效的非贵金属催化剂尤为必要[4-6]。CeO2具有萤石矿型结构,根据氧化还原气氛的不同,Ce可以在Ce4+/Ce3+之间转换,在结构中易于形成流动的氧空位,具有较好的储氧能力和氧化还原性能[7]。但CeO2易高温烧结,在应用于低温催化氧化CO时其储氧能力受到严重抑制[8-9]。研究表明,选择性暴露CeO2晶面对催化剂的性能具有较大影响[10-15],此外,通过元素掺加也能改善催化剂的性能。Mn具有多种价态,是氧化还原反应中常见的活性组分,Mn-Ce复合催化剂具有良好的储氧和氧化还原性能,是目前低温催化氧化CO的研究热点[16-20]。
本研究以Mn(NO3)2和Ce(NO3)3为原料,采用水热法制备了MnaCe1-aOx(a=0.01,0.02,0.05,0.08,0.10)纳米棒催化剂,运用TEM、XPS、XRD、ICP等手段进行了表征,并考察了Mn的掺加量对催化剂结构、活性、水热稳定性和催化氧化CO性能的影响。
1 实验部分
1.1 试剂和仪器
Mn(NO3)2溶液(w=50%)、Ce(NO3)3·6H2O、NaOH:分析纯。实验用水为去离子水。
D8-Advance型X射线衍射仪:德国Bruker公司;JW-BK112型比表面测试仪:北京精微高博科技公司;Tecnai G2 F20 S-TWIN型电子显微镜:美国FEI公司;AtomScan-16型电感耦合等离子体发射光谱仪:美国TJA公司;EScalab Xi+型X射线光电子能谱仪:美国Thermo Fisher公司。
1.2 MnaCe1-aOx催化剂的制备
MnaCe1-aOx催化剂的制备步骤为:1)称取1.3 g Ce(NO3)3·6H2O,再按n(Mn)∶n(Ce)为1∶99,1∶49,1∶19,2∶23,1∶9量取一定量的Mn(NO3)2溶液,将二者溶解于20 mL去离子水中,得到溶液Ⅰ;2)将14.4 g NaOH溶解于40 mL去离子水中,得到溶液Ⅱ;3)将溶液Ⅰ、Ⅱ混合,置于磁力搅拌器上,在室温下搅拌30 min;4)将混合后的溶液倒入容积为100 mL的高压反应釜(聚四氟乙烯内衬)中,在100 ℃条件下水热反应24 h后,冷却至室温取出,离心洗涤至上清液呈中性;5)将洗涤后的沉淀在80 ℃条件下干燥后置于马弗炉中,在500 ℃条件下焙烧 6 h,制得MnaCe1-aOx催化剂。
C e O2纳米棒(C e O2-N R)的制备步骤与MnaCe1-aOx相同,区别在于:制备过程中不添加Mn(NO3)2溶液。
将制得的CeO2-NR、MnaCe1-aOx催化剂在750 ℃、水蒸气体积分数为10%的气氛中老化10 h,得到高温水热老化催化剂,记作CeO2-NR HT、MnaCe1-aOxHT。
1.3 催化剂的表征
采用XRD表征催化剂的晶相结构;采用BET法测试催化剂的比表面积等物性参数;采用TEM观察催化剂的微观形貌结构及颗粒大小;采用ICP分析催化剂中金属元素的含量;采用XPS测试催化剂的表面元素和化学形态。
1.4 催化剂性能测试
催化剂性能测试在常压固定床反应器中进行。将0.4 g催化剂(40~60 目)固定在石英管中,用热电偶检测并控制催化床的反应温度,反应器出口配有气相色谱仪,对出口尾气中的CO进行在线检测[21]。反应开始前,在石英管内通入流量为100 mL/min的N2,在450 ℃条件下持续1 h排出其他气体,避免这些气体吸附在催化剂表面和孔道内对测试结果造成影响;随后通入反应气CO(φ=0.5%)持续1 h;在气体总流量为100 mL/min、体积空速为35 000 h-1的条件下,以N2作平衡气,通入反应气 CO(φ=0.5%)和O2(φ=0.25%);反应温度从50 ℃开始,以2 ℃/min的速率逐渐升温,每升温25 ℃采样(采样间隔为5 min,采样数量为5~6个,采样时长共30 min),直至CO转化率达到100%后停止升温。
2 结果与讨论
2.1 催化剂的活性评价
2.1.1 MnaCe1-aOx催化剂
以MnaCe1-aOx为催化剂时CO转化率随反应温度的变化见图1。通常将某组分转化率为50%时的反应温度(T50)作为该组分的起燃温度,转化率为90%时的反应温度(T90)作为该组分的完全转化温度[22]。由图1可见:CeO2-NR作催化剂时,T50和T90分别为230 ℃和 300 ℃;Mn0.01Ce0.99Ox作催化剂时,T50和T90都降低了约50 ℃,分别为180 ℃ 和250 ℃,表明掺加Mn后,催化剂的活性有明显提高;继续增大Mn的掺加量,Mn0.05Ce0.95Ox作催化剂时,T50和T90分别为160 ℃ 和192 ℃;进一步增大Mn的掺加量,Mn0.1Ce0.9Ox作催化剂时,T50和T90分别为120 ℃ 和160 ℃。
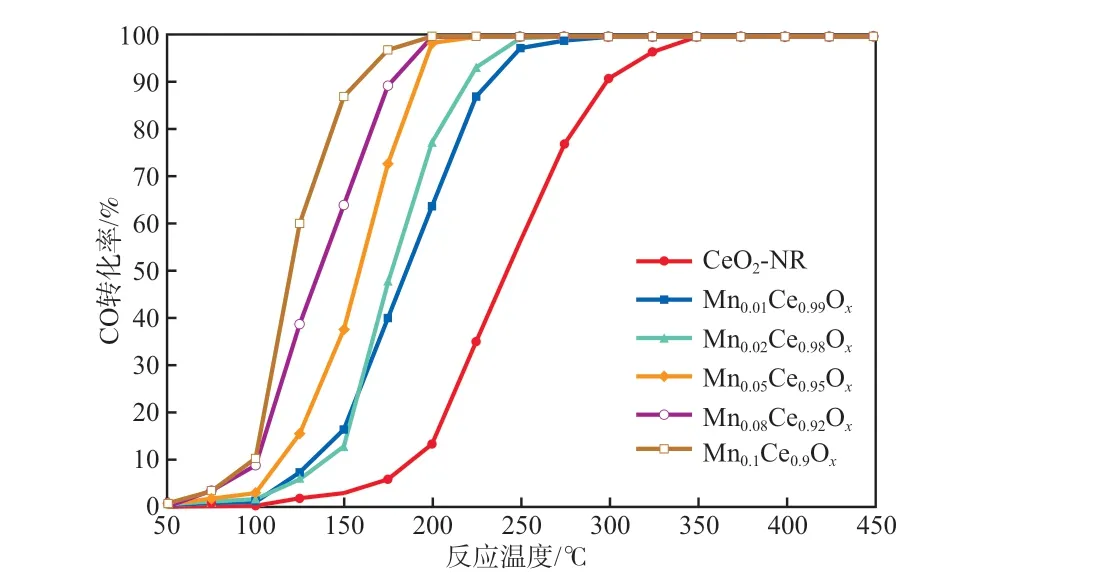
图1 以MnaCe1-aOx为催化剂时CO转化率随反应温度的变化
以MnaCe1-aOxHT为催化剂时CO转化率随反应温度的变化见图2。由图2可见:经高温水热老化后,CeO2-NR HT的活性明显下降,T90从300 ℃升高到350 ℃,这是因为高温烧结导致CeO2储氧性能下降,活性降低[23];掺加少量Mn后,MnaCe1-aOxHT的活性明显提高,Mn0.05Ce0.95OxHT作催化剂时,T90为175 ℃,原因可能是经高温水热处理后,Mn0.05Ce0.95OxHT形成了固溶体,存在Mn-O-Ce的强相互作用,从而使其抗水热性能显著提高;继续增加Mn的掺加量,MnaCe1-aOxHT的活性降低,Mn0.1Ce0.9OxHT作催化剂时,T90升高至185 ℃,其原因可能是催化剂中Mn掺加量过多导致晶格严重变形从而造成水热稳定性变差[24-25]。综上,确定Mn0.05Ce0.95OxHT为最适宜催化剂。
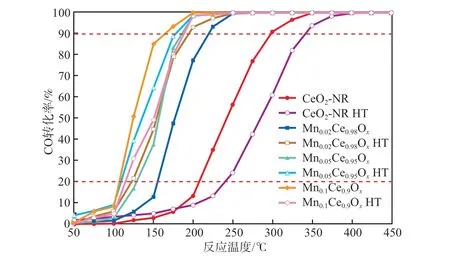
图2 以MnaCe1-aOx HT为催化剂时CO转化率随反应温度的变化
2.1.2 Mn0.05Ce0.95OxHT催化剂
在反应温度为175 ℃的条件下,考察了以Mn0.05Ce0.95OxHT为催化剂时CO转化率随反应时间的变化,结果见图3。由图3可见:在0~10 h内,CO转化率基本保持在89%左右,表明Mn0.05Ce0.95OxHT具有较高的催化活性;反应10 h后,在反应器中通入体积分数为10%的水蒸气,随着反应时间的延长,CO转化率缓慢下降,当反应时间为16 h时,CO转化率降低至86%,这可能是由于水蒸气和CO分子的竞争吸附导致催化剂活性下降;反应16 h后,停止通入水蒸气,CO转化率开始逐渐恢复,当反应时间为18 h时,CO转化率恢复到89%,进一步延长反应时间至30 h,CO转化率上升至92%左右,表明水蒸气对Mn0.05Ce0.95OxHT活性的抑制作用是可逆的,该催化剂具有良好的水热稳定性。
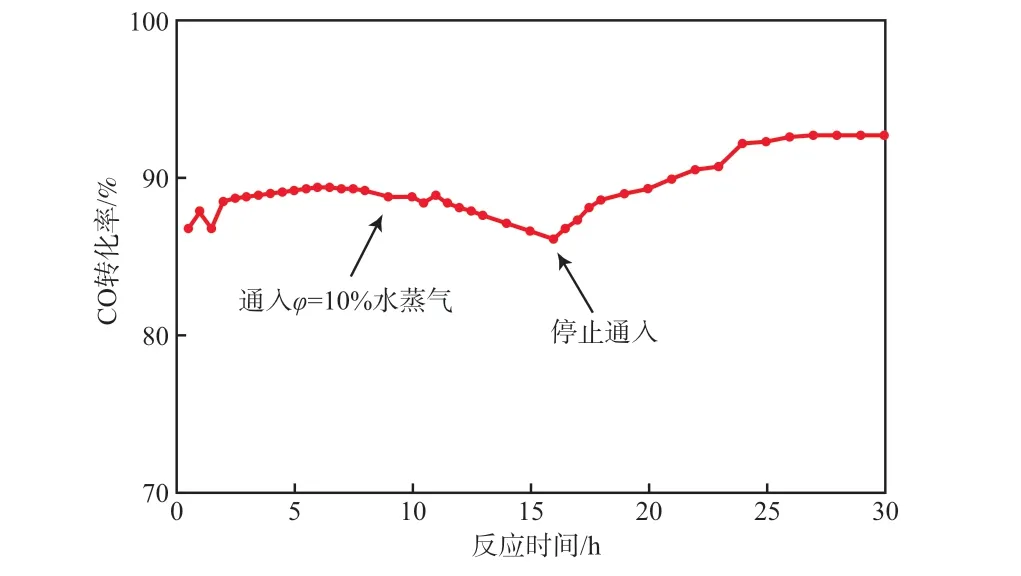
图3 以Mn0.05Ce0.95Ox HT为催化剂时CO转化率随反应时间的变化
2.2 MnaCe1-aOx催化剂的表征
2.2.1 TEM和ICP分析
图4为CeO2-NR和Mn0.05Ce0.95Ox的TEM和HRTEM照片。由图4a、4c可见:CeO2-NR是由具有均匀直径(约 14.3 nm)和不均匀长度(10~50 nm)的纳米棒构成;其晶面间距为0.26 nm,归属于(100)晶面[26];由图4b、4d可见,Mn0.05Ce0.95Ox的形貌特征与CeO2-NR相似,表明掺加少量的Mn元素并没有改变催化剂的形貌。

图4 CeO2-NR和Mn0.05Ce0.95Ox的TEM(a,b)、HRTEM(c,d)照片
通过ICP表征确定MnaCe1-aOx中Mn的实际摩尔分数,结果见表1。由表1可见,Mn摩尔分数的实测值与最初的投料比非常接近,表明Mn成功掺加进催化剂中。
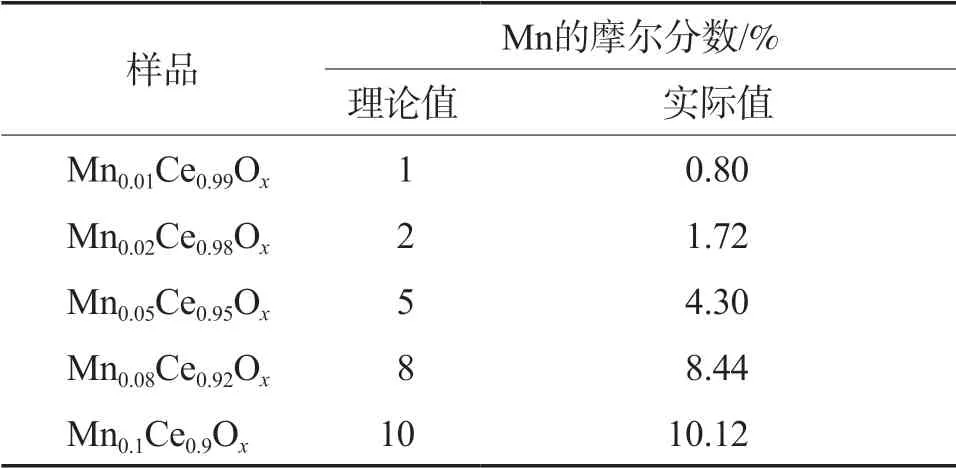
表1 MnaCe1-aOx 催化剂中Mn的摩尔分数
2.2.2 BET分析
MnaCe1-aOx的物性特征参数见表2。由表2可见:MnaCe1-aOx的比表面积为95~102 m2/g,较大的比表面积能够为CO的吸附提供有利条件;掺加Mn后,催化剂的比表面积变化不大,再次表明少量Mn的掺加不会破坏催化剂的结构;MnaCe1-aOx的孔体积为9.3~14.0 cm3/g,其中Mn0.05Ce0.95Ox的孔体积最大,为14.004 cm3/g,较大的孔体积有利于CO分子的扩散,进而有利于催化反应的进行;MnaCe1-aOx的粒径小于CeO2-NR,表明Mn的掺加能够阻止催化剂中晶粒的长大,从而提高其抗烧结能力[27];与MnaCe1-aOx相比,经水热处理后,MnaCe1-aOxHT的比表面积和孔径减小、粒径增大,这可能是由于催化剂中晶粒的团聚造成的。

表2 MnaCe1-aOx的物性特征参数
2.2.3 XRD分析
图5为Mn0.05Ce0.95Ox的XRD谱图。由图5可见:CeO2-NR在2θ为28.5°,33.1°,47.4°,56.3°,59.1°,69.4°处出现特征峰,依次对应于立方晶相CeO2(JCPDS,34-0394)的(111),(200),(220),(311),(222),(400)晶面;与CeO2-NR相比,掺加Mn后,Mn0.05Ce0.95Ox的XRD谱图未发生明显变化,表明Mn的掺加并没有改变催化剂的晶型;Mn0.05Ce0.95Ox的XRD谱图中未出现与MnOx有关的特征峰,可能是由于Mn的掺加量较少或者Mnx+进入了CeO2晶格,形成锰铈固溶体;与CeO2-NR相比,掺加Mn后,2θ为 28.5°的(111)晶面衍射峰向高角度方向偏移,这是由于Mnx+的离子半径(0.025~0.067 nm)小于Ce4+的离子半径(0.097 nm)[28],Mn掺加进CeO2的晶格导致晶格间距缩小。
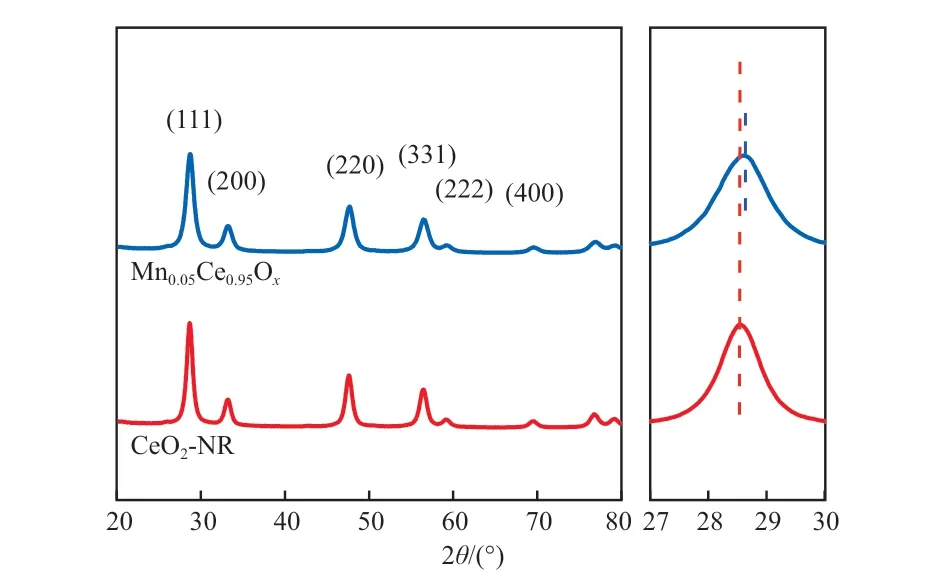
图5 Mn0.05Ce0.95Ox的XRD谱图
图6为Mn0.05Ce0.95OxHT的XRD谱图。由图6可见:与Mn0.05Ce0.95Ox相比,Mn0.05Ce0.95OxHT的衍射峰无明显偏移,表明水热处理并未引起催化剂晶格的变化;但Mn0.05Ce0.95OxHT的衍射峰强度明显增强,半峰宽缩小,表明水热处理后催化剂的结晶度明显提高。
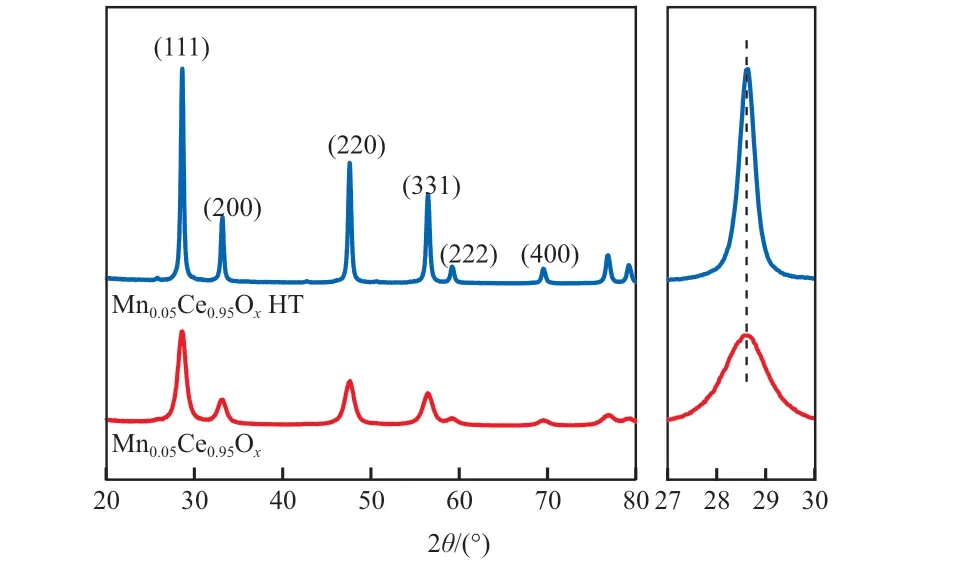
图6 Mn0.05Ce0.95Ox HT的XRD谱图
2.2.4 XPS分析
图7为MnaCe1-aOx中Ce 3d的XPS谱图。
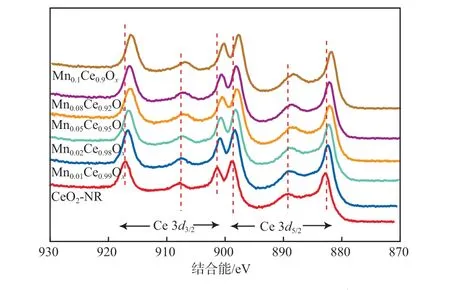
图7 MnaCe1-aOx 的Ce 3d 的XPS谱图
由图7可见:随着Mn掺加量的增加,MnaCe1-aOx中Ce 3d峰向高结合能方向偏移。
图8为MnaCe1-aOx和MnaCe1-aOxHT中Ce 3d的XPS谱图。由图8可见:Ce 3d的XPS谱图可以分解为8个峰,与4对自旋轨道双峰相对应。v,v′,v′,v′属于Ce 3d5/2,u,u′,u′,u′属于Ce 3d3/2;v,v′,v′′′,u,u′′和u′′归属于Ce4+特征峰,v′和u′归属于Ce3+特征峰,Ce3+和Ce4+在催化剂表面共存[29];水热处理对Ce 3d峰无明显影响。
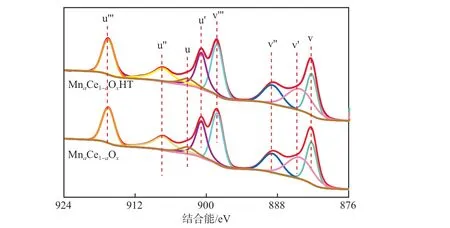
图8 MnaCe1-aOx和MnaCe1-aOx HT中Ce 3d 的XPS谱图
图9为MnaCe1-aOx中Mn 2p的XPS谱图。由图9可见:Mn 2p由Mn 2p1/2和Mn 2p3/2的自旋轨道双重态组成;与Mn3O4(641.3~641.4 eV)、Mn2O3(641.3~641.7 eV)和MnO2(641.7~642.2 eV)[30]相比,催化剂中Mn 2p3/2结合能(640.1~641.0 eV)有所降低,可能是由于锰氧化物高分散后与载体的相互作用增强,改变表面Mn物种的电子状态所致;随着Mn掺加量的增加,MnaCe1-aOx的Mn 2p峰向高结合能方向偏移0.2~0.8 eV,说明Mn的掺加使原子的化学状态发生了变化;Mn0.05Ce0.95Ox的Mn 2p3/2峰位于640.0,641.6,645.5 eV,分别归属于Mn3+、Mn4+和Mn3+表面粒子的振荡卫星峰。
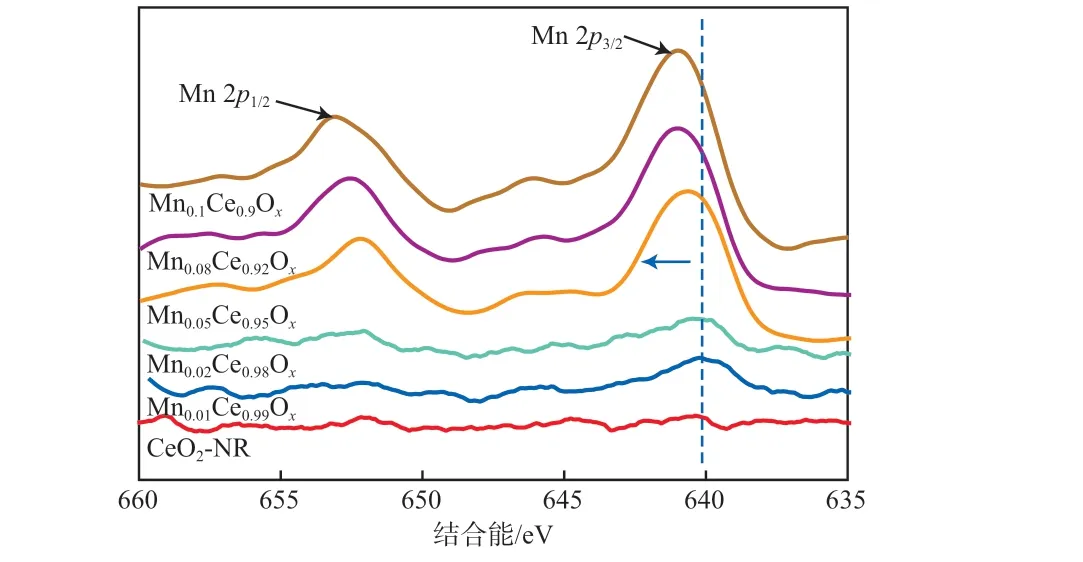
图9 MnaCe1-aOx中Mn 2p的XPS谱图
图10为MnaCe1-aOx和MnaCe1-aOxHT中Mn 2p的XPS谱图。由图10可见:经水热处理后,MnaCe1-aOxHT的Mn 2p峰向高结合能方向偏移了约0.8 eV,说明水热处理后部分Mnx+价态发生改变。
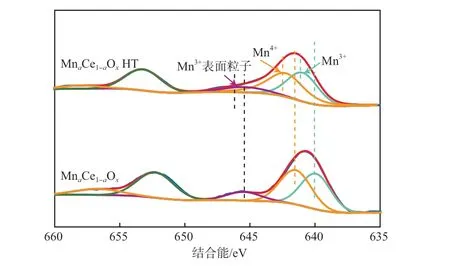
图10 MnaCe1-aOx和MnaCe1-aOx HT中Mn 2p的XPS谱图
3 结论
a)采用水热法制备了MnaCe1-aOx纳米棒催化剂,表征结果显示,Mn成功掺加进CeO2晶格,Mn0.05Ce0.95Ox催化剂为具有规则形貌的纳米棒,且选择性暴露面为(100)晶面。
b)Mn0.05Ce0.95OxHT的催化活性和水热稳定性均优于未掺加Mn的CeO2-NR HT;Mn0.05Ce0.95OxHT催化氧化CO时,T90为175 ℃,且稳定性较好。
c)Mn0.05Ce0.95OxHT催化活性提高的原因可能是由于Mn—O—Ce相互作用增强,催化剂中Ce3+的含量增加,晶格氧的流动性增强。
猜你喜欢
火炸药学报(2022年5期)2022-11-04
当代作家(2021年11期)2021-12-17
科学与财富(2021年33期)2021-05-10
科学(2020年4期)2020-11-26
科学(2020年4期)2020-01-11
数学物理学报(2019年5期)2019-11-29
物理实验(2019年7期)2019-08-06
航空材料学报(2019年2期)2019-04-15
汽车零部件(2018年5期)2018-06-13
云南民族大学学报(自然科学版)(2015年4期)2015-11-14
