
基于固相萃取-响应面优化的碳骨架在线催化/GC-MS/MS技术测定含油金属原料表面痕量氯污染物
2023-01-05 11:16张子豪郑建国麦晓霞彭速标刘莹峰
分析测试学报 2022年12期
张子豪,郑建国,麦晓霞,肖 前,彭速标,刘莹峰,李 丹
(广州海关技术中心,广东 广州 510623)
长期以来,我国金属冶炼行业面临节能减排的环保压力和高品质原料不足的问题,从国外进口相对短缺的金属废物原料成为解决我国金属资源不足的有效方案。2018年到2021年,我国数次调整《进口废物管理目录》,进口金属废物原料陆续被列入《禁止进口固体废物目录》[1]。由于国内金属冶炼产业对原料的需求依然旺盛,进口国外金属资源可以满足国内的部分原料需求,因此从国外进口高品质再生金属原料成为新趋势。但进口金属再生料可能存在品质参差不齐、夹杂物超标、有害物质超标等环保风险,其中粉尘、污泥、纤维等夹杂物易被识别与检测,而含油金属再生料中的油污特别是油污中含氯污染物因基质干扰大、目标物异构体多、含量较低等原因而难以准确测定。油污中含氯污染物(氯化石蜡、多氯联苯、多氯萘等物质)的普遍存在使其受到行业越来越多的关注。此类物质也是监管部门的重点关注对象。我国于2019年至2020年发布实施了GB/T 39733-2020《再生钢铁原料》、GB/T 38472-2019《再生铸造铝合金原料》、GB/T 38471-2019《再生铜原料》等系列再生金属标准[2-4]。其中规定再生金属原料中危险废物的质量不应超过总质量的0.01%。
再生金属原料主要由废旧金属通过简单拆解切割、筛分、冲洗后获得,可能会残留原设备工作环境或处理过程中加入的绝缘油、液压油、切削油等。其中可能含有亚老哥尔(工业多氯联苯)、卤蜡(工业多氯萘)、氯化石蜡(短链和中链)等耐热、阻燃、增塑类物质[5]。目前国内外对多氯联苯、多氯萘、氯化石蜡等物质的常规检测技术均有一些报道,如采用气相色谱-电子捕获检测器测定食品包装材料和皮革中的短链氯化石蜡[6-7],气相色谱-高分辨质谱法测定生物环境样品中的多氯联苯与多氯萘[8-9],气相色谱-质谱法测定玩具及泥土样品中的短链氯化石蜡与多氯联苯[10-12],液相色谱或质谱法测定食品和塑料等样品中的氯化石蜡、多氯联苯等[13-16],然而针对再生金属原料中含氯污染物的测定和在线催化技术的应用未见报道。含氯有机污染物为人工合成物质,主要由对应的芳烃与脂肪族正构烷烃氯化制得,由于碳骨架氯化位置及氯化度等均存在差异,使有机氯化物存在上百种异构体[17]。实际测定过程中目标物结构复杂,缺少对应的标准品,多氯联苯混合物特别是氯化石蜡混合物的分离效果不理想,峰形均为宽谱带的峰簇,当目标物共同存在时峰形均有一定叠加,特征离子也可能部分重叠而难以识别,导致灵敏度下降等均是此类物质难以准确定量的重要原因。
本研究以超声浸渍法合成的Pd/改性活性炭为催化剂,将目标物在富氢环境下在线脱氯加氢,含氯污染物转化为特征直链烷烃和芳烃,使测定目标从多氯烃分散的同位素离子转换为集中的高响应烷烃、芳烃离子,进一步减少干扰,提高检出限,并利用气相色谱-串联质谱多反应监测模式测定。该方法灵敏度高、准确稳定,适用于金属原料中多氯联苯(PCBs)、多氯萘(PCNs)、短链和中链氯化石蜡(SCCPs、MSCCPs)等含氯物质的快速筛查和测定。
1 实验部分
1.1 仪器与试剂
Agilent 7890B-7000C气相色谱-质谱/质谱仪(美国Agilent公司);Precision Hydrogen 100氢气发生器(英国Peak公司);AS20500A超声波发生器(加拿大AUTO Science公司);可调移液器(1~100 μL,德国普兰德公司);DB-5MS UI石英毛细管柱(30 m×0.25 mm,0.25 μm,美国Agilent公司);不分流单锥衬管(容量900 μL,内径4 mm,美国Agilent公司);脱活玻璃棉(色谱级,美国Agilent公司);活性炭粉(200目,上海麦克林生化科技有限公司);自制固相萃取柱(硝酸银硅胶固相萃取柱:1 g/6 mL)[18]。
C10~C17正构烷烃(正癸烷、正十一烷、正十二烷、正十三烷、正十四烷、正十五烷、正十六烷和正十七烷)、四氢萘(KDTN)、苯基环己烷(HCB)、正戊基苯、一氯代烃(1-氯癸烷、1-氯十一烷、1-氯十二烷、1-氯十三烷、1-氯十四烷、1-氯十六烷、1-氯萘和2-氯联苯)、1,1,1,3,11,13,13,13-八氯十三烷、八氯萘、2,2,3,4,5-五氯联苯、2,2,3,3,4,4,5,5,6-九氯联苯、短链氯化石蜡(100 μg/mL,平均氯化度51.5%)、中链氯化石蜡(100 μg/mL,平均氯化度52.0%)、多氯萘1014(100 μg/mL,平均氯化度63.5%)和多氯联苯1260(100 μg/mL,平均氯化度60.0%)标准物质均购自德国Dr.Ehrenstorfer GmbH公司,纯度≥98%。正己烷、二氯甲烷(HPLC级,美国Tedia公司);无水乙醚(分析纯,99%,广州化学试剂厂);氯化钯(99.9%,广州市丹安仪器仪表有限公司);冰醋酸(99.5%,广州化学试剂厂);氨水(分析纯,广州化学试剂厂)。
1.2 标准工作溶液的配制
称量一定质量的上述还原产物(C10~C17正构烷烃、四氢萘、苯基环己烷)和一氯代烃(1-氯癸烷、1-氯十一烷、1-氯十二烷、1-氯十三烷、1-氯十四烷、1-氯十六烷、1-氯萘和2-氯联苯)共18个标准物质于18个10 mL棕色容量瓶中,用正己烷稀释定容,配制质量浓度均为1 000 μg/mL的还原产物标准储备液和一氯代烃标准储备液。内标储备液:称量一定量的正戊基苯于10 mL棕色容量瓶中,用正己烷配制1 000 μg/mL的内标标准储备液。工作溶液:使用还原产物标准储备液和内标储备液,用正己烷配制目标物质量浓度均为0.02、0.05、0.10、0.25、0.50、1.00 μg/mL的还原产物标准工作溶液,内标质量浓度为0.25 μg/mL。使用一氯代烃标准储备液和内标储备液,配制质量浓度为1.00 μg/mL的一氯代烃标准工作溶液,内标质量浓度为0.25 μg/mL。催化效率质控溶液:使用短链氯化石蜡(氯化度为51.5%)、中链氯化石蜡(氯化度为52.0%)、多氯萘1014和多氯联苯1260标准溶液配制各物质质量浓度均为0.50 μg/mL的质控溶液,内标质量浓度为0.25 μg/mL。
1.3 样品预处理
称取50.0 g(精确至0.01 g)金属原料碎料样品于250 mL旋盖瓶中,加入150 mL正己烷,40℃下超声萃取30 min,溶液转移至150 mL平底烧瓶,旋蒸浓缩至15 mL后转移至25 mL容量瓶中,正己烷定容。移取以上溶液1 mL至已用0.5 mL正己烷活化的硝酸银硅胶固相萃取柱中,待充分吸收后加入3.0 mL正己烷-乙醚(体积比9∶1)混合溶液,当溶液全部通过萃取柱筛板后,弃去以上淋洗液,更换接收瓶。继续向固相萃取柱中加入2.5 mL正己烷-乙醚(1∶1)混合溶液,收集上述洗脱液,氮吹至干后,用配有内标(质量浓度为0.25 μg/mL)的正己烷溶液定容至1 mL,过滤后待测定。
1.4 催化剂的制备和反应衬管的填装
称取10 g活性炭加入150 mL煮沸后的10%硝酸水溶液中,搅拌处理30 min,用去离子水多次冲洗至中性后过滤,得到的活性炭用去离子水超声10 min,过滤并在60℃下干燥得到改性活性炭。
采用超声辅助浸渍法制备Pd/改性活性炭催化剂前驱体[7],称取84 mg固体氯化钯粉末于50 mL烧杯中,加入20 mL 0.06 mol/L盐酸溶液,45℃搅拌溶解,加入5 g改性活性炭和1.5 g聚乙二醇分散,超声20 min后静置。将上述溶液置于95℃干燥箱中干燥得到黑色粉末,即为Pd/改性活性炭催化剂前驱体。
取不分流单锥衬管,底部填入脱活玻璃棉后,从下往上依次加入1 mm高度的碳酸钙和10 mm高度的Pd/改性活性炭催化剂前驱体,最后覆盖一层玻璃棉,将该衬管装入气相色谱进样口,320℃下以氢气为载气活化1 h,即得Pd/改性活性炭催化剂反应衬管。
1.5 气相色谱-串联质谱条件
色谱柱:DB-5MS UI石英毛细管柱(30 m×0.25 mm,0.25 μm);载气:高纯氢气;碰撞气:高纯氮气;柱流速:1.5 mL/min;进样口温度:260℃;分流比:不分流;起始温度:45℃(保持3 min),以15℃/min升至250℃(保持1 min),最后以30℃/min快速升至300℃。
电子轰击离子源(EI),能量:70 eV;传输线温度:300℃;离子源温度:230℃;四极杆温度:150℃;多反应监测(MRM)负离子模式,溶剂延迟5 min。
2 结果与讨论
2.1 净化与洗脱条件的优化
非极性溶剂可将样品洗出液中的非极性杂质(如矿物油、切削油中脂肪族烃等物质)去除,实验采用硝酸银硅胶固相萃取柱作为净化柱,分别以环己烷、正己烷、正戊烷、二氯甲烷4种溶剂作为淋洗溶剂。配制含有正十二烷、正十四烷、四氢萘、苯基环己烷、短链氯化石蜡(氯化度51.5%)和多氯联苯1260质量浓度均为1.00 μg/mL的混合标准溶液,分别用1.0、1.5、2.0、2.5、3.0 mL的淋洗溶剂对上述标准溶液进行回收率实验。结果表明,以相同体积正己烷或正戊烷淋洗时,正构烷烃的回收率较高,四氢萘和苯基环己烷的回收率较低;环己烷与正己烷的淋洗效果相似,四氢萘和苯基环己烷的回收率略有改善;二氯甲烷由于极性较大,易将正构烷烃、四氢萘、苯基环己烷与少量氯化石蜡和多氯联苯一起洗出,因此单一溶液均不适合作为淋洗溶剂。选用正己烷-乙醚(9∶1)和正己烷-二氯甲烷(9∶1)混合溶剂分别作为净化淋洗溶剂进行实验,结果表明,正己烷-二氯甲烷(9∶1)的洗脱能力稍强。这是由于二氯甲烷相对于乙醚的溶解范围较大,溶解性好。但当苯基环己烷完全洗出时伴有少量氯化石蜡被同步带出,因此选择正己烷-乙醚(9∶1)混合溶液作为淋洗溶剂。考察了不同体积淋洗溶剂对目标化合物回收率的影响,使用2.0 mL淋洗溶剂时,正构烷烃已完全洗出;3.0 mL淋洗溶剂时,四氢萘和苯基环己烷的回收率达到96%以上;淋洗溶剂达3.5 mL以上时有少量短链氯化石蜡和多氯联苯被洗出,因此选择3.0 mL正己烷-乙醚(9∶1)混合溶剂作为淋洗溶剂。
硝酸银硅胶柱分离饱和烃、芳烃类与氯代化合物的效果较好,这可能归因于银离子与目标物氯原子形成的弱络合作用,从而实现了对含氯烃较强的保留。样品经正己烷-乙醚(9∶1)淋洗净化后,萃取柱上的含氯化合物需用合适的洗脱溶剂洗脱出来。氯化烷烃和芳烃类物质因碳骨架上氯化位置不同而具有一定极性,可被二氯甲烷、酯类、醚类等溶剂洗脱,因此选择极性不同的二氯甲烷-乙醇(5∶1)、二氯甲烷、正己烷-二氯甲烷(1∶1)、正己烷-乙醚(1∶1)、正己烷-乙酸乙酯(1∶1)作为洗脱溶剂。选择不同体积(1.5、2.0、2.5、3.0、3.5 mL)的洗脱溶剂,用上述一氯代烃标准工作溶液和催化效率质控溶液(混合氯代烃)进行回收率实验,每个条件进行2次实验。结果表明,加入相同体积的洗脱溶剂时,前2种溶剂的洗脱效果均较理想,回收率达95%以上,但溶剂极性较强,洗脱出样品中芳烃类和酯类物质,可能会对芳烃和直链烷烃类还原产物造成干扰。正己烷-乙酸乙酯混合溶剂的洗脱效果一般,平均回收率约在85%左右;正己烷-乙醚和正己烷-二氯甲烷混合溶剂对氯化石蜡和多氯联苯类物质的洗脱效果均较好,洗脱体积在2.5 mL以上时回收率达到95%,但混合溶剂中含有过多的二氯甲烷可能会影响后续催化反应的效率。综合考虑,采用2.5 mL正己烷-乙醚(1∶1)混合溶液作为洗脱溶剂。
2.2 催化剂的选择
分别采用水浴浸渍法制备Pd/玻璃珠载体催化剂[7]、超声辅助浸渍法制备Pd/改性活性炭催化剂、Pt-Sn/氧化铝催化剂[19]和Pt-Ag/活性炭催化剂[20]。以上4种催化剂均在320℃下用氢气活化1 h,活化后保持干燥。采用填有上述4种催化剂的反应衬管,对1-氯十三烷和八氯十三烷标准溶液进行脱氯加氢催化反应,对比脱氯催化效果。实验表明,上述2种氯代烃在4种不同的催化衬管中均能转化为对应的还原产物。双金属催化体系中,Pd-Ag/活性炭催化剂因Ag的加入催化活性有所降低,还原产物的转化率较低,高氯化合物产物中还含有部分未充分还原的低氯化合物,如三氯十三烷。Pt-Sn/氧化铝催化剂的催化活性较高,能对烯烃和酮类物质产生加成反应,增加了样品分析的复杂性。Pd/玻璃珠载体催化剂和Pd/改性活性炭催化剂的催化效果适中,其中以改性活性炭为载体的催化效率更高,这可能与活性炭经硝酸预处理和超声辅助浸渍处理有关。硝酸的氧化作用使活性炭表面的氧官能团增加而增强了其吸附能力,加之超声辅助也能更好提高活性炭载体的吸附性能,使活性金属Pd能均匀地分布在活性炭的内外表面,因此催化效率更高。最终选择Pd/改性活性炭催化剂作为脱氯加氢催化剂。图1为氢气活化后的Pd/改性活性炭催化剂与Pd/改性活性炭催化剂前驱体的二维X射线衍射谱图对比,活化后催化剂在2θ=40°左右处有1个较小的钯衍射峰,说明还原产生了金属钯,并附着于改性活性炭上。
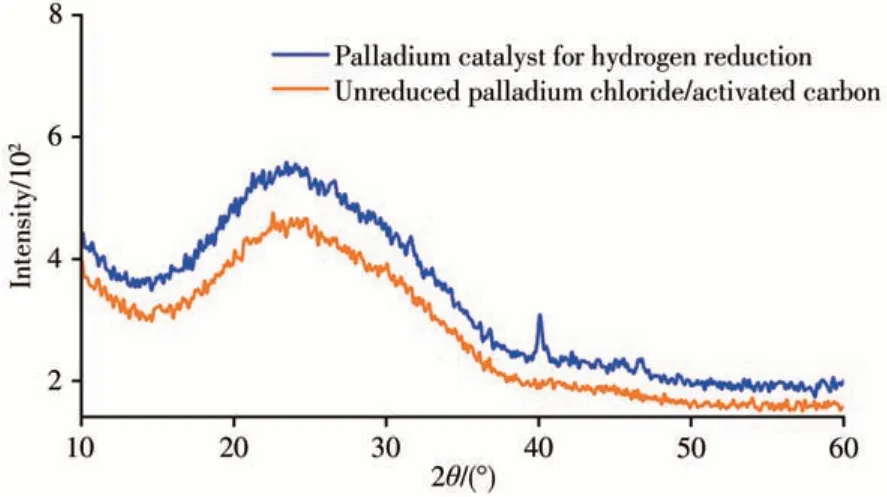
图1 Pd/改性活性炭催化剂的二维X射线衍射谱图Fig.1 Two-dimensional X-ray diffraction patterns of Pd/activated carbon catalyst
2.3 硝酸浓度对催化活性的影响
本实验发现,经硝酸改性的活性炭载体钯催化剂的催化活性优于未经硝酸改性的活性炭载体钯催化剂。这是因为经硝酸氧化改性后,活性炭表面的含氧官能团数量显著增加,含氧官能团通过氢键、共价键和其他物质相互作用,可提高活性炭的吸附能力。实验考察了质量分数分别为5%、10%、15%、20%、25%的硝酸水溶液对活性炭改性后催化活性的影响。结果表明,用10%硝酸改性处理的活性炭催化效果最好,硝酸浓度大于10%后,催化活性逐渐降低。这可能是由于高浓度的硝酸改性处理活性炭时降低了载体的比表面积,导致催化活性下降,最终选择10%硝酸为活性炭改性用硝酸的浓度。
2.4 催化衬管条件的优化
2.4.1 温度对催化效率的影响反应衬管中催化剂在氢气环境中活化,以一氯代烃标准工作溶液为研究对象,考察了进样口温度(200、220、240、260、280、290、300、310℃)对催化效率的影响,以进样口温度对各一氯代烃还原产物(C10~C14及C16饱和直链烃、苯基环己烷和四氢萘)的色谱响应作图(如图2所示)。结果表明,8种一氯代烃在不同温度下还原,氯代脂肪烃产物的响应峰面积随温度升高而升高,在240~280℃左右峰面积最高,温度高于290℃,峰面积有所下降。氯代芳烃的还原现象与氯代脂肪烃有所区别,氯代芳烃随催化温度的变化会生成不同的还原产物。如多氯联苯与多氯萘的还原产物较复杂,多氯联苯、多氯萘除了能够脱氯加氢转变为联苯和萘,还会进一步还原生成苯基环己烷、四氢萘和少量甘菊环烃类物质,多氯萘和多氯联苯在200~280℃范围内的主要还原产物为四氢萘和苯基环己烷,还原产物的峰面积与温度成反比,温度升高会抑制还原产物四氢萘和苯基环己烷的生成。温度高于270~280℃左右,还原产物萘和联苯的含量上升,四氢萘和苯基环己烷的含量下降。

图2 温度对催化性能的影响Fig.2 Effect of temperature on catalytic performance
2.4.2 氢气流速对催化效率的影响载气作为还原氢的来源使样品中的氯代烃在氢气氛围下催化还原。氢气流速对催化剂的催化效率有一定影响,通过设置不同的氢气流速(1.0、1.2、1.4、1.6、1.8 mL/min)对1-氯萘和2-氯联苯的标准溶液进行测试。结果显示,氢气流速从1.0 mL/min升至1.8 mL/min时,还原产物的保留时间逐渐变小,峰面积缓慢变大。这可能是由于在相对较高的氢气流速下,进样口压力有所上升,目标物质与催化剂的接触更加充分。流速为1.4~1.6 mL/min时,催化效率趋于稳定。
2.4.3 氯化程度对催化效率的影响选取1-氯十三烷(MCTRID)、1,1,1,3,11,13,13,13-八氯十三烷(OCTRID)、1-氯萘(MCN)、八氯萘(OCN)、2-氯联苯(PCB1)、2,2,3,4,5-五氯联苯(PCB87)和2,2,3,3,4,4,5,5,6-九氯联苯(PCB206)储备液,配制成质量浓度均为10 μg/mL的工作溶液,在“1.5”条件下进行脱氯加氢还原实验,催化效率如表1所示。结果表明,分子中氯的质量分数越低,还原得到的烃类物质丰度越大,峰面积越大。7种化合物的催化效率均不低于95.1%,可以看出催化剂对一氯代烃和多氯代烃的催化效率无明显差别,说明氯含量对催化剂的催化效率影响较小。

表1 不同氯含量的一氯代烃和多氯代烃的催化效率Table 1 Catalytic efficiencies of chlorinated hydrocarbons with different chlorine contents
2.4.4 催化参数的优化脂肪族氯代烃和芳香族氯代烃的脱氯规律不同,因此需对催化条件进行优化,以1-氯十三烷和2-氯联苯为脂肪族氯代烃和芳香族氯代烃的代表物质,采用响应面模型确认最优参数。氯化程度对催化效率的基本影响较小,因此选取催化过程中影响较大的催化温度(A,℃)、氢气流速(B,mL/min)和可能对催化效率有影响的化合物质量浓度(C,μg/mL)、催化剂用量(D,g)4个参数作为变量,以某金属原料加标样品进行实验,每一变量设置3个水平,试液中1-氯十三烷和2-氯联苯的催化效率为响应值,根据Box-Behnken中心组合实验设计原理,分别进行4因素3水平共29次实验。得到四因素交互影响显著的1-氯十三烷和2-氯联苯的催化效率响应面图如图3所示,氯代脂肪烃的回归模型方程为:catalytic efficiency=88.40+4.42A-0.92B-0.67C+0.50D+1.50AB+0.75AC+1.00AD+1.75BC-0.50BD-12.99A2-1.74B2-0.12C2-2.12D2;氯代芳烃的回归模型方程为:catalytic efficiency=73.60-14.67A+2.25B-1.00C+1.75D+1.75AB+0.50AC-0.25AD-0.25BC-4.74BD-3.25CD-7.34A2+0.03B2+0.66C2-2.47D2。

图3 四因素交互影响显著的1-氯十三烷和2-氯联苯的催化效率响应面图Fig.3 Response surface and contour of the recoveries for MCTRID and PCB1 affected alterately by four factors
对上述实验结果进行方差分析,由表2可知该模型的F值为6.75和19.76,模型概率p值均小于0.05,说明各影响因子对催化效率的影响显著。通过软件拟合得到调整相关系数分别为0.871 0和0.951 8,模型拟合度良好,说明相关性较好,能解析模型87.10%和95.18%的响应值变化,可以此模型对再生金属原料中氯代烃的催化效率进行分析。由两个模型的F值可知该模型一次项A的影响均极显著,C的影响不显著。单因素对脂肪族氯代烃回收率的影响顺序为A>B>C>D,芳香族氯代烃为A>B>D>C,催化温度最为显著,氢气流速次之。脂肪族氯代烃的二次项A2、B2、D2影响显著,C2影响不显著;芳香族氯代烃的二次项A2、D2影响显著,C2和B2影响不显著。结合实际样品处理和催化效率分析,经响应面模型分析得出最佳参数:催化温度为260℃,氢气流速为1.5 mL/min、催化剂用量为2 g。为进一步验证数据的可靠性,采用上述最优条件对已知浓度的1-氯十三烷和2-氯联苯加标混合溶液进行催化实验,重复3次,得到催化效率分别为87.3%和92.1%,与理论值相当,所以最终采用上述条件进行实验。

表2 Box-Behnken中心组合设计的方差分析结果Table 2 Analysis of variance results for Box-Behnken central composite design
2.5 质谱条件的选择
将几种氯代烃还原产物的标准溶液逐个在EI全扫描模式下以相同的电离能量进行一级质谱扫描。结果表明,C10~C17直链烃类物质均较易形成分子离子峰[M]+,如[C13H28]+(M=184.3)和[C16H34]+(M=226.1);也易于形成相差若干个—CH2—的 系 列 离 子 峰,如[C3H7]+(M=43.1)、[C4H9]+(M=57.3)、[C5H11]+(M=71.2)等。芳烃类物质易形成分子离子峰[M]+,如[C10H12]+(四氢萘,M=132.1)和[C12H16]+(苯基环己烷,M=160.1)等分子离子峰。分别对以上各分子离子以0~60 eV的碰撞能量进行产物离子模式扫描,结果表明,直链烃类物质分子离子均易形成响应最强的烷烃特征峰,[C4H9]+(M=57.3)或[C5H11]+(M=71.2)的分子离子峰,此类分子离子峰均能进一步丢失—CH2—形成分子量更低的[M-14]+烷烃或烯烃特征峰,如[C3H7]+(M=43.1)、[C3H5]+(M=41.1)和[C2H3]+(M=27.1)的分子离子;芳烃类物质易形成带有烃基取代的苯环结构特征峰,如[C9H9]+(α-甲基苯乙烯,M=116.9)、[C8H8]+(苯乙烯,M=104.2)、[C7H7]+(甲苯,M=91.1)、[C6H5]+(苯,M=77.1)的分子离子峰。几种氯代烃还原产物和内标物正戊基苯优化后的参数见表3,MRM总离子流色谱图见图4。

图4 还原产物混合标准溶液(1.0 μg/mL)的MRM色谱图Fig.4 MRM chromatogram of reduction products mixed standard solution(1.0 μg/mL)the peak numbers denoted were the same as those in Table 3
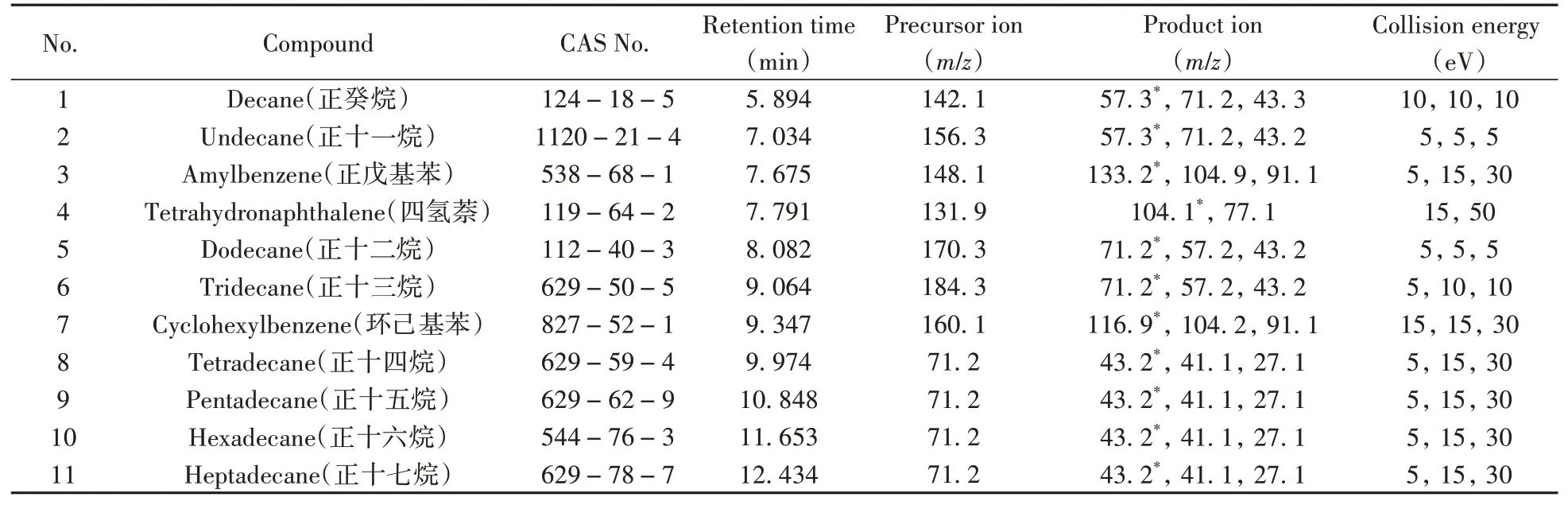
表3 还原产物及内标物优化后的定量、定性离子对和碰撞能量Table 3 Optimized parameters of quantitative ion,qualitative ion and collision energy for reduction products
2.6 线性范围与定量下限
氯代烃目标物通过在线催化,转化为C10~C17直链烷烃、四氢萘和苯基环己烷而被测定。用“1.2”的催化效率质控溶液作为测试催化剂转化效率的质控标样,使用还原产物标准工作溶液,以各还原产物与内标物的质量浓度比(X)和各还原产物与内标物的色谱峰面积比(Y)绘制标准工作曲线。结果表明,10种还原产物在0.02~1.00 μg/mL质量浓度范围内线性良好,相关系数(r2)为0.995 1~0.999 3。以10倍信噪比(S/N)作为定量下限(LOQ),得到还原产物的定量下限为0.011~0.040 μg/mL,即还原产物C10~C13直链烷烃总和为0.098 μg/mL,C14~C17直链烷烃总和为0.124 μg/mL,四氢萘为0.012 μg/mL,苯基环己烷为0.011 μg/mL(见表4)。方法定量下限相当于样品中短链氯化石蜡的含量为4.07 μg/kg、中链氯化石蜡的含量为5.33 μg/kg、多氯萘的含量为0.29 μg/kg、多氯联苯的含量为0.26 μg/kg。
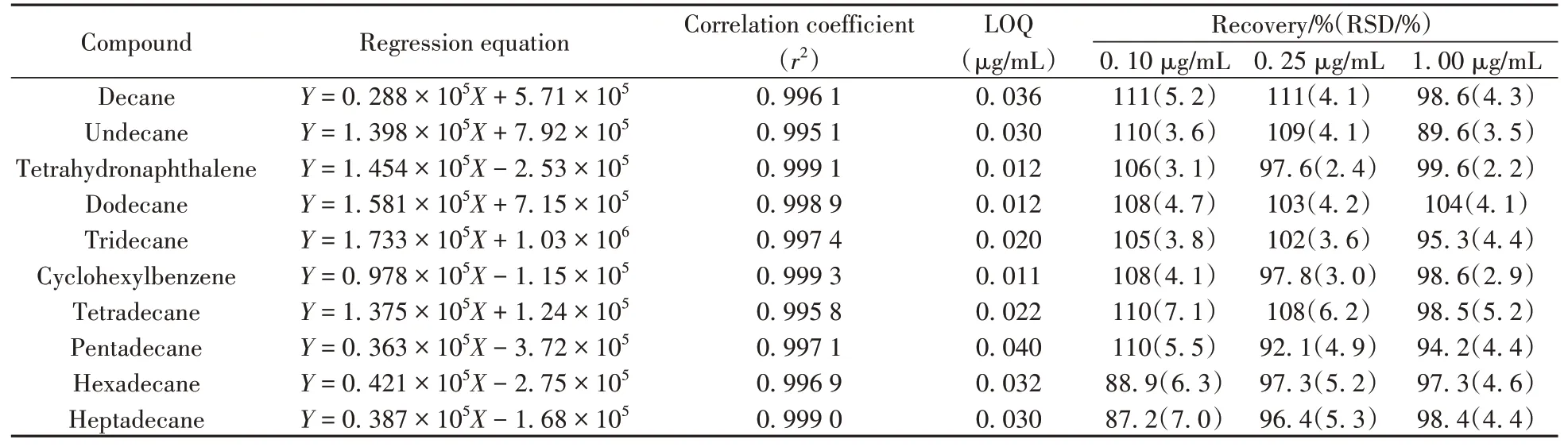
表4 还原产物的回归方程、相关系数、定量下限、回收率和相对标准偏差Table 4 Regression equations,correlation coefficients,limits of quantitation,recoveries and relative standard deviations of the reduction products
2.7 回收率与相对标准偏差
选取不含C10~C17直链氯代烷烃(短链及中链氯化石蜡)、多氯萘和多氯联苯的含油金属原料,向其中加入适量短链氯化石蜡、中链氯化石蜡、多氯萘1014和多氯联苯1260标准溶液,配制成目标物质量浓度均为0.10、0.25、1.00 μg/mL的基质样液,在催化剂催化效率90%以上时测试上述3个浓度基质样液中还原产物的浓度,每个加标水平平行测试6次,计算回收率和相对标准偏差(RSD)。由表4可知,样品的加标回收率为87.2%~111%,RSD不大于7.1%。
2.8 实际样品的测试
采用本方法对10批次进口含油金属原料样品中短链氯化石蜡、中链氯化石蜡、多氯联苯和多氯萘的含量进行测定,每个样品平行测试2次。结果表明,2个样品检出短链氯化石蜡、中链氯化石蜡和多氯联苯。1#样品中检出SCCPs:152.6 μg/kg,MSCCPs:38.4 μg/kg,PCBs:0.9 μg/kg;2#样品中检 出SCCPs:19.3 μg/kg,MSCCPs:44.8 μg/kg,PCBs:39.4 μg/kg。某含油金属原料样品的MRM色谱图见图5。
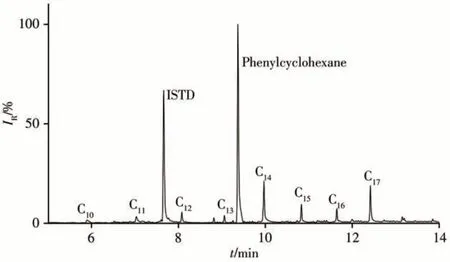
图5 某含油金属原料样品的MRM色谱图Fig.5 MRM chromatogram of an oil-bearing metal raw material
3 结论
本文运用氯代烃钯催化技术,采用催化衬管通过在线催化/气相色谱-串联质谱法测定进口含油金属原料中多氯联苯、多氯萘、短链和中链氯化石蜡等含氯污染物的含量。目标物碳骨架通过脱氯加氢,使含氯异构体混合物转变成C10~C17正构烷烃、四氢萘和苯基环己烷,利用气相色谱-串联质谱MRM模式内标法进行测定。方法减少了多氯类物质各同系物之间的干扰,从而实现了含油金属原料中氯污染物的准确测定。该方法较为简便、灵敏度高,能满足进口含油再生金属原料中含氯污染物的批量筛查和检测需求。
猜你喜欢
化工设计(2022年4期)2023-01-02
科学导报(2022年28期)2022-05-24
石油炼制与化工(2022年2期)2022-02-15
化工管理(2021年7期)2021-05-13
化工管理(2020年26期)2020-10-09
环境保护与循环经济(2020年4期)2020-06-08
中国特种设备安全(2019年1期)2019-03-13
分析化学(2018年12期)2018-01-22
电子技术与软件工程(2016年24期)2017-02-23
分析化学(2014年11期)2014-11-19
