
超高效液相色谱-四极杆-飞行时间质谱技术非靶向定性筛查猪肉中79种药物残留
2023-01-05 11:16王建凤高丽娟王秋水冯月超
分析测试学报 2022年12期
刘 佳,王建凤,高丽娟,王秋水,冯月超,刘 艳,丁 奇,王 颖,邵 鹏
(北京市科学技术研究院分析测试研究所(北京市理化分析测试中心),北京 100089)
现代畜牧业中,兽药被广泛用于维持动物健康、预防感染和治疗疾病[1]。然而,用药不规范或者非法使用违禁药物将导致动物源性产品中违禁药物检出、兽药残留超过国家标准规定,产生食品安全风险[1-4]。为了确保食品安全,国际食品法典委员会、欧盟委员会、中国等均规定了食品中药物最大残留限量[5],如GB 31650-2019[6]《食品安全国家标准-食品中兽药最大残留限量》中规定了动物源性食品中磺胺类抗生素、倍氯米松等的最大残留限量。考虑到养殖者用药的不确定性和复杂性,建立一种高通量、快速测定动物源性食品中多药物残留的检测方法刻不容缓。
动物源性食品存在基质复杂、干扰物多等特点,且多药物分析中药物极性往往差异较大,单一的前处理技术难以适用于所有目标物。最初用于农药残留分析的快速样品前处理技术——QuEChERS目前已被广泛用于动物源性食品中多类兽药的残留分析[7-9],然而该方法无法充分回收动物养殖中常用的极性药物,如四环素和喹诺酮类[5]。因此,建立一种适用于多组分的前处理技术十分必要。
目前,液相色谱-串联质谱法被广泛用于动物源性食品中多药物的同时检测[7,10-13]。该法具有高的灵敏度和选择性,但存在两点不足:一是无法分析多反应监测(MRM)中未定义的化合物,因此无法获得关于未知药物的信息[14];另一方面,通过串联质谱进行多药物残留分析可能非常耗时,尤其是对数百种不同化合物的分析[15]。四极杆飞行时间质谱(TOF MS)具有数据采集速度快、分辨能力高、质量精度高、检测灵敏度高等多重优点,能够同时分析无限数量的化合物,实现非靶向目标化合物定性,近年来已发展成为一种高效的筛查方法,在食品安全分析领域有害物质高通量、快速筛查方面得到广泛应用[9,14,16-19]。
本研究以GB 31650-2019[6]、《中华人民共和国农业农村部公告第250号公告》[20]等中限用、禁用的β-兴奋剂、蛋白同化激素、抗生素等79种药物为研究对象,在优化样品前处理技术的基础上,采用超高效液相色谱-四极杆-飞行时间质谱(UPLC-Q-TOF MS)技术,建立了猪肉中79种药物的多残留定性筛查方法。该方法具有通量高、简便、快速等特点,具有较强的实际应用价值。
1 实验部分
1.1 仪器、试剂与样品
Acquity UPLC超高效液相色谱仪、SYNAPT G2-Si四极杆飞行时间质谱仪、Waters Acquity BEH HSS-C18柱(美国Waters公司);GR22GⅢ高速冷冻离心机(日本Hitachi公司);MS200多管涡旋混匀仪(杭州瑞诚仪器有限公司);N-EVAPTM112恒温水浴氮吹仪(美国Organomation Associates公司);Secura225D-1CN天平(德国Sartorius公司);Vortex Genius 3旋涡混合器(德国IKA公司);Milli-Q超纯水仪(美国Millipore公司);FAVEX-NM50兽药残留快速柱(500 mg/6 mL,高雄巨研科技股份有限公司)。
79种药物标准品(纯度≥98%)购自德国Dr.Ehrenstorfer公司和美国A Chemtek公司;乙腈、甲醇、甲酸(色谱纯)、甲酸铵(美国Thermo Fisher Scientific有限公司);叔丁基甲基醚(色谱纯,美国Sigma-Aldrich公司);碳酸钠(Na2CO3)、乙二胺四乙酸二钠盐(EDTA-Na2)(分析纯,国药集团化学试剂有限公司);0.22 μm有机尼龙滤膜(天津津腾公司)。
所用样品为市售猪肉鲜肉,购自当地超市。
1.2 实验方法
1.2.1 溶液配制
根据各药物标准品的溶解性,选择甲醇、乙腈等溶剂配制质量浓度100~1 000 μg/mL的各药品单一标准品储备液,-18℃避光保存。
混合标准储备液:分别移取适量单一标准品储备液于10 mL容量瓶中,用甲醇稀释配制成1 μg/mL的混合标准储备液。
基质标准工作溶液:取适量混合标准储备液,用基质空白提取液逐级稀释,配制成质量浓度分别为0.01、0.05、0.1、0.25、0.5、1、2、5、10、20、50、100、200 μg/L的基质标准工作溶液,现用现配。
1.2.2 样品制备
1.2.2 .1样品提取称取粉碎均质后的样品2 g(精确至0.01 g)于50 mL聚丙烯离心管中,先加入10 mL 0.5%(体积分数)甲酸-乙腈溶液,充分混匀后以2 500 r/min振荡提取30 min,10 000 r/min下冷冻离心10 min后将上清液全部移出,再加入10 mL甲醇,以2 500 r/min振荡提取30 min,10 000 r/min冷冻离心10 min,上清液全部移出。分别准确移取两次上清液各5 mL,置于FAVEX-NM50净化柱中,以每秒1滴流速加压,收集滤液,于40℃氮吹至近干,用1 mL乙腈(0.1%甲酸)∶5 mmol/L甲酸铵(0.1%甲酸)(13∶87,体积比)定容,涡旋复溶,经0.22 μm有机尼龙滤膜过滤,待仪器分析。
1.2.2 .2基质空白提取液的制备取不含被测物的猪肉样品,按照“1.2.2.1”操作处理,得到基质空白提取液。
1.2.3 分析方法
(1)城乡发展联动性不足。长沙市城乡旅游产业结构和要素目前尚不完整,与工业、农业、林业以及其他产业融合不足:县域内旅行社、游客集散中心、旅游咨询中心等功能发挥的不够;餐饮住宿、休闲经营规模有待扩大,服务水平有待提升;缺少规模化的特色手工艺品、土特产等旅游商品及其加工的销售场所。同时由于各镇村各自为政、争先恐后地做规划、出台优惠办法来发展乡村旅游,统筹开发不足、景点同质化情况日渐增多。
1.2.3 .1色谱条件色谱柱:Waters Acquity BEH HSS-C18(2.1 mm×150 mm,1.7 μm);流动相A:0.1%(体积分数)甲酸-乙腈;流动相B:0.1%(体积分数)甲酸-5 mmol/L甲酸铵溶液。梯度洗脱程序:0~0.5 min,13%A;0.5~10.0 min,13%~50%A;10.0~10.75 min,50%~95%A;10.75~12.25 min,95%A;12.25~12.5 min,95%~13%A;12.5~15.0 min,13%A。柱温:40℃;进样量:10 μL;流速:0.4 mL/min。
1.2.3 .2质谱条件离子源:电喷雾离子源(ESI);扫描方式:正离子扫描;离子源温度:150℃;电喷雾电压:3 000 V;脱溶剂气流速:800 L/h;脱溶剂气温度:400℃;锥孔气流速:50 L/h;锥孔电压:15 V;扫描时间:0~15 min;扫描间隔时间:0.1 s;MSE(全信息串联质谱)模式检测,灵敏度模式;质量扫描范围:m/z50~1 200;数据采集模式:Continuum模式;碰撞能量范围:10~60 eV。质量校正液为亮氨酸脑啡肽。
1.2.4 数据处理
利用Masslynx软件对前处理后的样品进行数据采集,以UNIFI软件进行数据分析,基于实验室自行建立的包括79种药物在内的质谱数据库[21]进行检索,以目标化合物的一级质谱精确分子量、保留时间、碎片离子等信息进行药物残留的快速定性分析。
2 结果与讨论
2.1 色谱条件优化
所测定的79种药物化学性质相差较大,较难达到良好分离,尤其是对于分子式相同或结构式相近的化合物,其有相近或相同的碎片离子,且出峰时间相近易造成谱图干扰。对于高分辨质谱,目标物质在色谱柱上的分离程度越高,其二级质谱的干扰碎片信息越少,更有利于定性结果的准确判断[22]。实验发现有机相为乙腈时,化合物峰形对称性好、分离度高、保留较好;当在水相和有机相中加入甲酸时,可明显提高β-兴奋剂、糖皮质激素、蛋白同化激素类化合物在离子源中的离子化效率,增加正离子目标物质的响应值。同时,甲酸铵溶液对部分目标物质的色谱峰形具有改善作用。因此,结合出峰时间、峰形以及化合物响应,选用乙腈(0.1%甲酸)-5 mmol/L甲酸铵(0.1%甲酸)作为流动相,梯度洗脱程序见“1.2.3.1”。
2.2 样品前处理条件优化
进行非靶向筛查时要求前处理方法能够全面地提取样品中的化合物[23]。因此,以提取物质的数量尽可能多为前提进行样品前处理条件优化,对提取溶剂进行考察。
2.2.1 提取溶剂优化
本研究中化合物的极性范围非常广泛,且绝大多数目标化合物在有机溶剂中的溶解度较大,因此分别采用甲醇、乙腈、叔丁基甲基醚中的一种或多种溶剂对目标化合物进行提取。已有研究表明[16,24],提取溶剂的pH值会影响酸碱两性物质的电离,因此,同时考察了有机溶液中加入弱酸和弱碱对目标物的提取效果。分别研究了①甲醇,②0.5%甲酸-甲醇,③乙腈,④0.5%甲酸-乙腈,⑤甲醇和乙腈等比例混合溶剂(含0.5%甲酸),⑥甲醇、乙腈和0.1%EDTA-Na2等比例混合溶剂(含0.5%甲酸),⑦10%Na2CO3和叔丁基甲基醚(1∶9,体积比)的提取效果。不同提取溶剂下目标化合物的提取情况如表1所示,提取液⑥和⑦分别提取出77种和51种目标化合物,除四环素类中的土霉素外,提取液③提取出其它78种目标化合物。其余4种提取溶剂均提取出全部79种目标化合物,目标物提取率达100%。在这4种提取溶剂中,79种目标化合物的提取回收率如图1所示。由于目标化合物性质差别较大,其回收率表现出较大差异:对于大环内酯类以及磺胺类抗生素,甲醇表现出明显优于其他3种提取溶剂的提取效果;0.5%甲酸-乙腈对大多数喹诺酮类抗生素、β-兴奋剂、蛋白同化激素、糖皮质激素以及孕激素拮抗剂的提取效果优于其它3种提取溶剂。综合考虑79种物质的平均回收率和目标化合物在不同回收率范围的个数(表2),选择0.5%甲酸-乙腈和甲醇组合作为提取溶剂,并对两种溶剂的组合顺序进行优化。
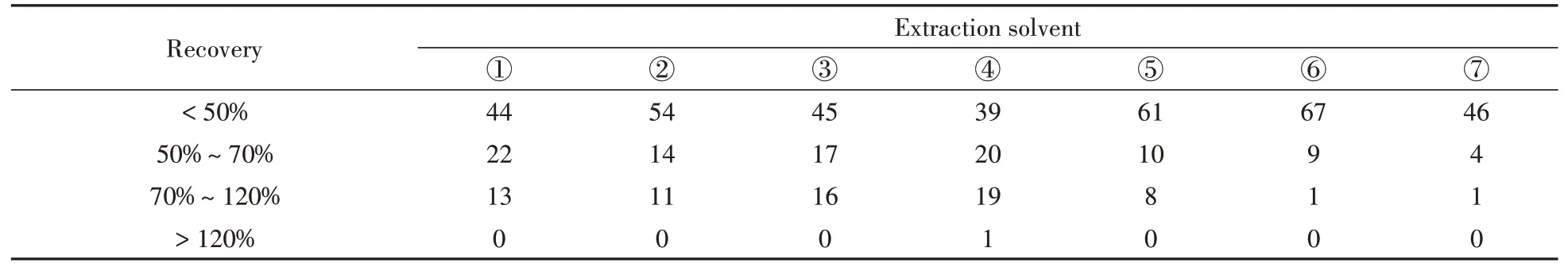
表2 不同提取溶剂下猪肉中79种目标化合物在不同回收率范围的个数统计Table 2 Statistics of 79 target compounds in pork under different extraction solvents in different recovery ranges

图1 不同提取溶剂对79种目标化合物回收率的影响Fig.1 Effect of different solvents on the recoveries of 79 target compounds
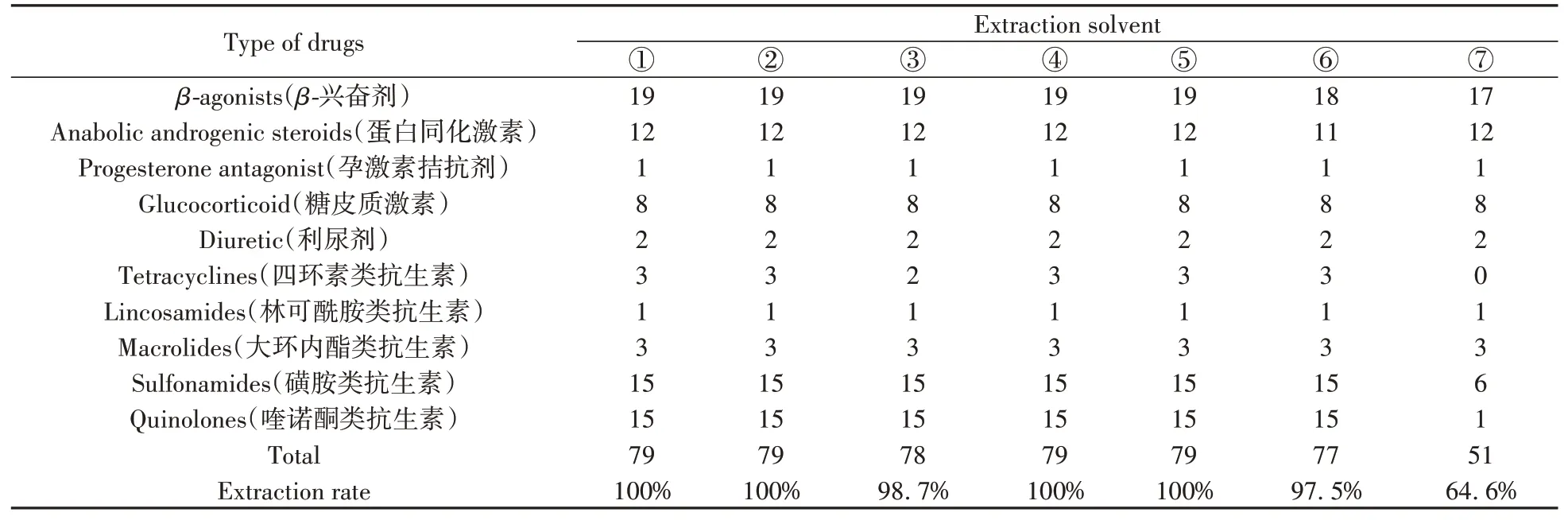
表1 不同提取溶剂对猪肉中79种目标化合物的提取情况Table 1 Extraction of 79 target compounds from pork with different extraction solvents
2.2.2 提取溶剂组合优化
由于物质种类较多,每类化合物中选择2~3种代表性物质,考察①先甲醇后0.5%甲酸-乙腈;②先0.5%甲酸-乙腈后甲醇;③甲醇和0.5%甲酸-乙腈同时提取对回收率的影响,结果如图2所示。除阿奇霉素、罗红霉素外,组合②对于选定目标化合物的提取效果优于其他两种组合。因此,采用先0.5%甲酸-乙腈后甲醇的组合提取顺序。

图2 部分目标化合物在不同组合提取顺序下的回收率Fig.2 Recoveries of some target compounds under different solvent combinations
2.3 基质效应
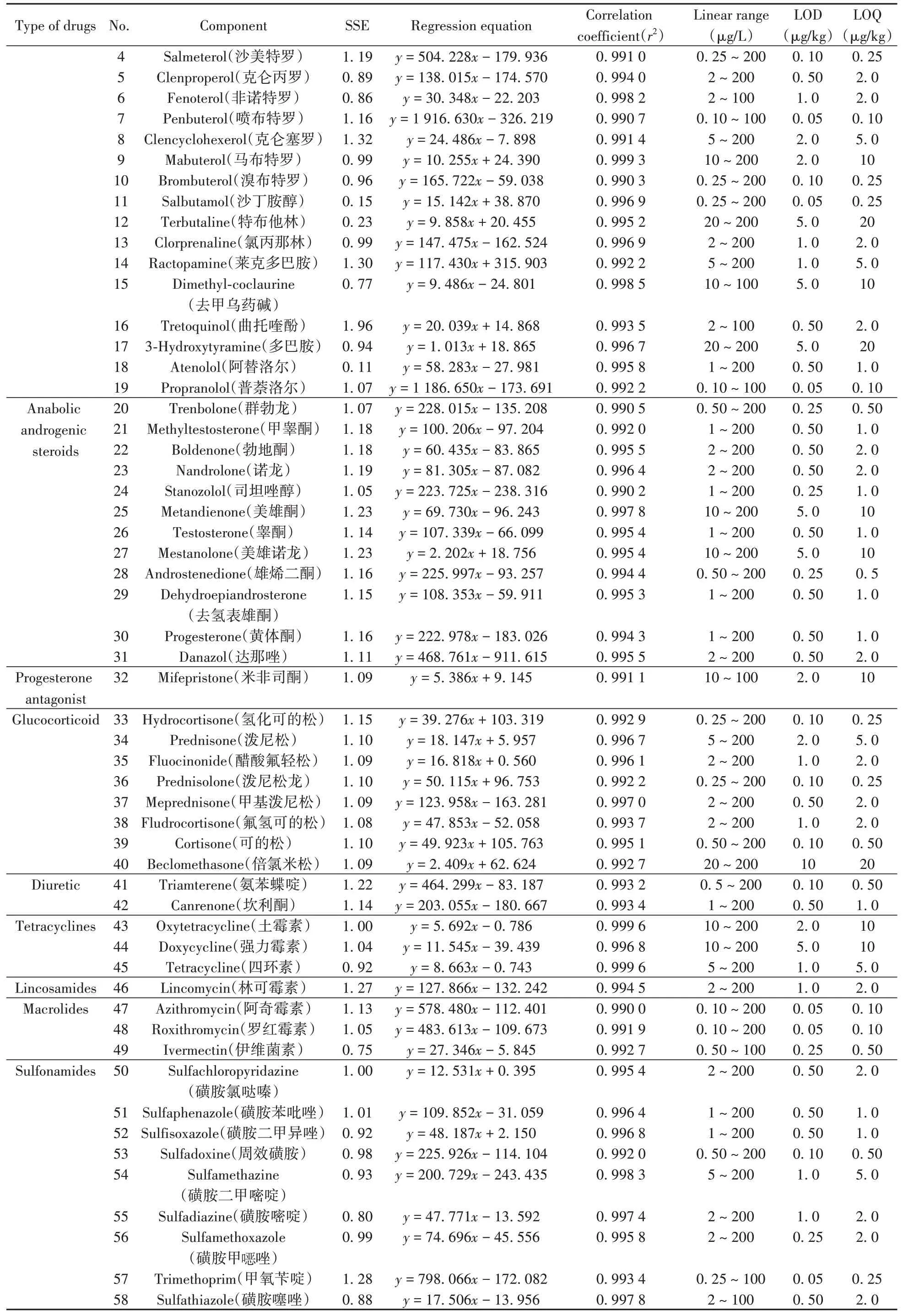
基质效应是指目标分析物以外的其他组分引起的分析信号增强或抑制现象[25],通过采用基质标准曲线斜率与溶剂标准曲线斜率的比值(SSE)来评价基质效应[26]。SSE值为0.8~1.2时,表明基质效应不明显[27];当SSE>1.2时,表明存在基质增强效应;当SSE<0.8时,表明存在基质抑制效应[28]。本实验分别配制猪肉空白基质标准曲线与对应浓度的溶剂标准曲线考察SSE值,结果见表3。结果表明:86.1%(68/79)的目标物质SEE在0.8~1.2范围内,6.3%(5/79)的目标物质SEE<0.8,存在基质抑制效应,7.6%(6/79)的目标物质存在基质增强效应。基质效应影响定量结果的准确性,通常采用同位素内标法或基质匹配标准曲线法[22]进行校正。本方法的目标物质接近百种,使用内标的成本偏高,不利于方法的推广,因此,采用空白基质标准曲线进行校正以降低基质对测定结果的影响。

表3 猪肉中79种药物的信号抑制/增强、线性方程、线性范围、相关系数、检出限和定量下限Table 3 Signal suppression/enhancements(SSEs),regression equations,linear ranges,correlation coefficients(r2),limits of detection(LODs)and limits of quantitation(LOQs)of 79 drugs in pork
(续表3)

(续表3)
2.4 线性关系、检出限及定量下限
用空白基质提取液配制系列浓度的79种药物混合标准工作溶液,以峰面积(y)为纵坐标,对应的质量浓度(x,μg/L)为横坐标,绘制基质标准工作曲线,79种药物在相应质量浓度范围内线性关系良好,相关系数(r2)均不低于0.99(表3)。分别以3倍信噪比(S/N)和10倍信噪比对应的浓度确定方法的检出限(LOD)和定量下限(LOQ)[28],79种药物的LOD为0.05~10 μg/kg,LOQ为0.10~20 μg/kg。通过与GB 31650-2019[6]中提及的相关药物残留限量相比,该方法定量下限远低于对应物质限量标准,可以很好地满足食品中药物残留的筛查需求。
2.5 非靶向筛查技术的应用
2.5.1 模拟加标样本分析
选取每类物质中的1~3种代表性物质共21种(普萘洛尔、群勃龙、勃地酮、诺龙、米非司酮、雄烯二酮、氢化可的松、泼尼松、去氢表雄酮、可的松、倍氯米松、氨苯蝶啶、阿奇霉素、罗红霉素、伊维菌素、磺胺二甲嘧啶、磺胺嘧啶、甲氧苄啶、磺胺甲噻二唑、氧氟沙星、奥比沙星),采用所建立的方法对加标水平为10 μg/kg和20 μg/kg的上述21种物质的猪肉样本进行检测。通过检索质谱信息数据库,采用精确质量数和保留时间的方式进行非靶向定性筛查,在质量数精确度允许偏差为10 ppm,保留时间偏差为0.2 min,且至少检测到1种碎片离子的条件下,10 μg/kg加标水平的筛查结果见表4,部分典型药物的全扫描图见图3。由表4可知,10 μg/kg加标下,除定量下限高于10 μg/kg的倍氯米松(表3)未筛查出外,其余物质全部筛出;20 μg/kg加标下21种物质可全部筛查出来。

图3 部分典型药物的全扫描图Fig.3 Full-scan chromatograms of some typical drugs

表4 10 μg/kg加标水平下样本的筛查结果Table 4 Screening results of specimen at 10 μg/kg spiked level

(续表4)
2.5.2 实际样本检测
采用本方法对市售的6份猪肉样本进行筛查分析,结果显示,除1份样本为阴性外,其余5份样本均检出氢化可的松,含量为1.39~13.6 μg/kg。所有阳性样本中氢化可的松的离子加和模式均为[M+H]+。为了检验筛查结果的准确性,采用高灵敏度Xevo TQ-S串联四极杆质谱[8]对阳性样本进行验证,以样品3为例,Xevo TQ-S串联四极杆质谱仪的确证色谱图见图4。与本方法结果一致。
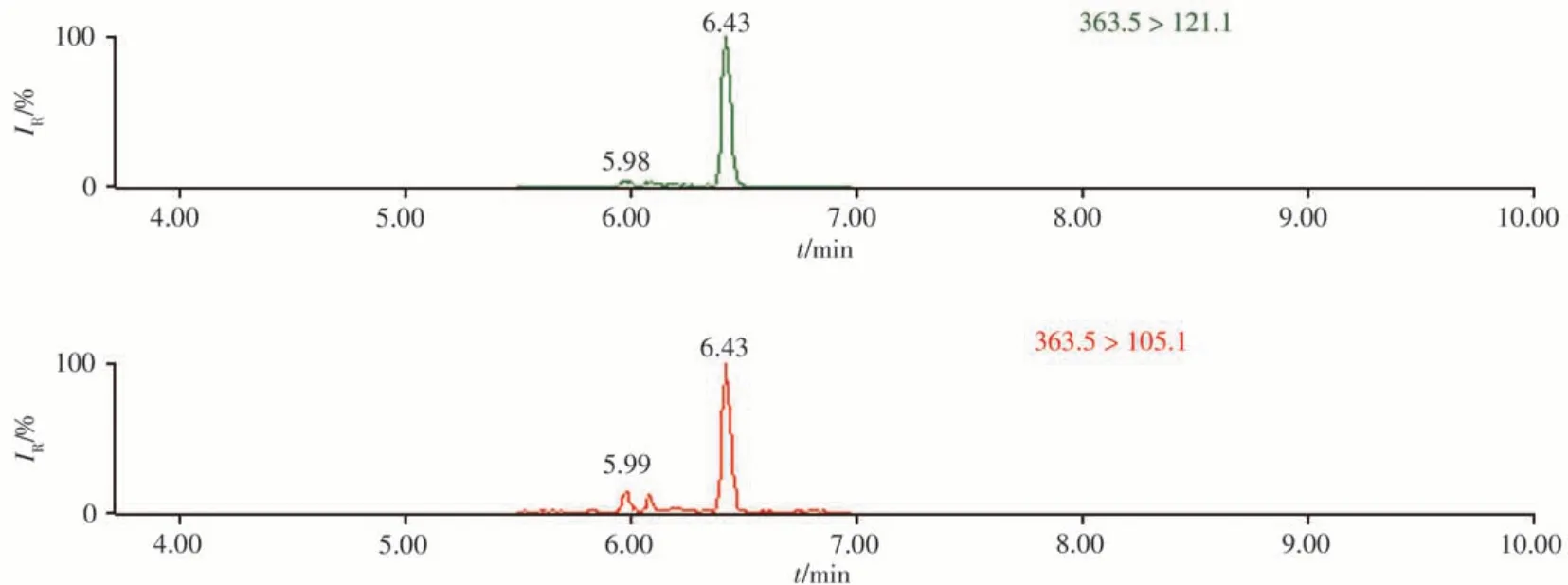
图4 样品3的Xevo TQ-S串联四极杆质谱仪色谱图Fig.4 Xevo TQ-S tandem quadrupole mass spectrometer chromatogram of sample 3
我国GB 31650-2019[6]中规定,氢化可的松是被允许外用于所有食品动物且不需要制定残留限量的兽药。结果表明6份市售猪肉样本在所研究的79种药物范围内是安全的。
3 结论
本研究基于UPLC-Q-TOF MS,建立了无需标准品即可实现猪肉中79种药物残留的快速定性筛查技术,并将其应用于模拟阳性样本以及随机市售样本的筛查。结果表明该方法具有通量高、简便、快速、高效等优点,可用于风险预警、日常监测及应急检测,为猪肉类食品安全提供了有力的技术支撑。
猜你喜欢
煤化工(2022年3期)2022-07-08
核化学与放射化学(2022年2期)2022-04-28
现代仪器与医疗(2022年1期)2022-04-19
合成纤维工业(2021年5期)2021-10-31
食品安全导刊(2021年20期)2021-08-30
现代仪器与医疗(2021年2期)2021-07-21
首都食品与医药(2020年1期)2020-10-21
灾害医学与救援(电子版)(2018年1期)2018-06-05
中国蜂业(2018年4期)2018-05-09
肥料与健康(2018年6期)2018-03-04
