
Selection of specif ic nanobodies against peanut allergen through unbiased immunization strategy and the developed immuno-assay
2023-01-23 09:14YozhongHuChunZhngJingLinYiWngSihoWuYingSunBoweiZhngHunXuemengJiYngLuShuoWng
食品科学与人类健康(英文) 2023年3期
Yozhong Hu, Chun Zhng, Jing Lin, Yi Wng, Siho Wu, Ying Sun,Bowei Zhng, Hun Lü, Xuemeng Ji, Yng Lu, Shuo Wng,*
a Tianjin Key Laboratory of Food Science and Health, School of Medicine, Nankai University, Tianjin 300071, China
b College of Food Science and Engineering, Tianjin University of Science and Technology, Tianjin 300457, China
Keywords:Peanut allergen Ara h 3 Nanobody Sandwich ELISA Gly 1
A B S T R A C T Peanut allergy is considered to be a major health issue with global effects. To date, no effective curative approach has been applied for the therapy of the anaphylaxis resulting from the peanut allergens. The accurate and effective detection methods for the surveillance of allergens in food are still the primary strategies to avoid allergic diseases. In this study, nanobodies (Nbs) derived from the Heavy-Chain only Antibodies(HCAbs) were selected against the general peanut protein extract through the unbiased strategy to facilitate the development of the sandwich ELISA for the detection and surveillance of peanut allergen contamination.The target antigen of the selected Nb was identified as peanut allergen Ara h 3, and a cross-reaction was observed with the member of Gly 1 from the Ara h 3 family. The applicability of the self-paired Nb P43 on the establishment of the immuno-assay was verif ied. A sandwich ELISA against peanut allergen was developed,which reached a linear range of 0.2-10.6 μg/mL, and a limit of detection of 53.13 ng/mL.
1. Introduction
Peanut is on the list of recommended daily intake since the abundant dietary component and nutritional value [1,2]. Whereas,the prevalence of peanut-originated allergen-induced allergic disease,especially the allergic reaction throughout the whole life, has drawn dramatic attention to the research about the allergen detection to avoid the unexpected intake of the peanut allergen, and the therapeutic approaches after intake [3,4]. Peanut-induced allergic disease has contributed to the highest mortality for allergic reactions [5-7]. So far, 17 peanut allergens (Ara h 1-17) have been designated by the International Federation of Immunologic Societies (IUIS) allergen Nomenclature Committee (http://www.allergen.org/search). Among them, Ara h 1, Ara h 2 and Ara h 3 are considered to be the most important peanut allergens [8,9]. It was demonstrated that Ara h 3 could be recognized by serum IgE from 40% of the peanut allergen sensitive population [10,11].
Currently, no effective therapeutic strategy has been applied for the anaphylaxis resulted from peanut allergens, and the sensitive detection and avoidance of the unexpected intake are still the prior strategy to prevent allergic reactions [12,13]. Therefore, the establishment of the peanut allergen detection method has become extremely important and applicable for allergen surveillance in food matrix. Various detection methods have been developed [14-16].Pomés et al. [17] reported a specific two-site sandwich ELISA to detect peanut Ara h 1 in chocolate in 2003. In 2004, Stephan et al. [18]developed a sandwich ELISA using polyclonal antibodies for the detection of trace peanut antigens in processed foods.
Traditionally, the preparation of specific antibodies depends on the purified allergen. The immunization with purified allergen is expected to raise enough immune response to facilitate the following selection of positive clones [19,20]. The immunization with general extract from peanut will be more cost-effective for the immunization process, whereas, the possibility to obtain specific antibodies is obscure,and is necessary to be determined. The current study was proposed to employ an alternative candidate of nanobodies (Nbs) to verify the research hypothesis and to establish an immuno-assay against peanut allergen, which should be feasible in view of the very straightforward selection strategy and evaluation protocol of Nbs [21,22].
The background of Nbs started from the report about a unique antibody format in camels coined Heavy-Chain only Antibody(HCAb) naturally devoid of the light chains and CH1 domain [23].The cloning of the antigen binding repertoire of HCAb could facilitate the engineered antibody fragment of VHH, also called nanobodies because of its size in nanometer range (2.4 × 4 nm) [24,25]. Because of its small size and complete antigen-binding capacity, Nbs has been considered as the ideal carrier in basic research, diagnosis and treatment of certain diseases [26-31]. The robust characteristics of Nbs including the high thermal stability and good solubility, high affinity and specificity against the target have facilitated the primary consideration in various research [32-34].
Herein, the current study is proposed to select specific Nbs against peanut allergen through the unbiased selection strategy by immunizing with general protein extract from peanuts. Generally, Nbs against peanut protein have been selected and confirmed with high specificity and affinity. More importantly, the target of one Nb has been verified as peanut allergen of Ara h 3, which directly provided the evidence for the successful application of Nbs on unbiased allergen analysis.Based on the selected Nb, an initial immunoassay of sandwich ELISA has been established for the detection of Ara h 3 with relatively high sensitivity and specificity. Moreover, the combination of Nbs with advanced detection techniques, such as fluorescence or biosensorbased immunoassay could further advance the detection of Ara h 3 allergen with high sensitivity and applicability.
2. Materials and methods
2.1 Material
LymphoprepTMDensity Gradient Medium were purchased form STEMCELL Technologies (Canada).E. coliTG1 competent cells were purchased from American Lucigen company. NaH2PO4were provided by Sinopharm Chemical Reagent. All the chemicals and reagents utilized in the study are of analytical grade unless otherwise indicated.
2.2 Preparation of peanut protein extract
Twenty grams of dry peanuts were ground and defatted using acetone(1:10,m/V) at 4 °C for 2 h. Centrifugation was performed at 8 000 r/min for 30 min to collect the precipitate for the following evaporation in the fume hood overnight. Crude extraction of peanut protein (1:5,m/V)was accomplished by using 50 mmol/L Tris-HCl buffer (containing 1.0 mol/L NaCl, pH 8.0) after stirring at 4 °C overnight. The supernatant was then collected after centrifugation to serve as the crude extract of peanut protein. Dialysis was performed for buffer exchange and the sample was concentrated by using an ultrafiltration tube. The protein extract was visualized by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) after Coomassie-blue staining. The concentration of peanut protein was determined by using the BCA protein concentration detection kit (Thermo Scientific).
2.3 Immunization and contraction of the library
A young alpaca was immunized with the crude peanut extract mixed with complete or incomplete Freund’s adjuvant to raise an immune response. Generally, the immunization was performed by 6 injections at weekly intervals, each with 0.5 mg protein. 50 mL of anti-coagulated blood was then collected from the jugular vein three days after the last boost. Peripheral blood lymphocytes were isolated with LymphoprepTMDensity Gradient Medium (STEMCELL Technologies) The total RNA was purified from prepared lymphocytes by TRIzolTMReagent (Invitrogen) and used for cDNA synthesis. Subsequently, cDNA was used as a template to amplify theVHHgene fragment by two steps of RT-PCR. In the first step of PCR amplification, CALL001 (5’-GTC CTG GCTGCT CTT CTA CAA GG-3’) and CALL002 (5’-GGT ACG TGC TGT TGA ACT GTT CC-3’) were used as primers to amplify fragments between the VHH and CH2 regions. The amplified products were subjected to agarose gel electrophoresis to extract the fragments corresponding to a size of around 700 bp by using QIAquick Gel Extraction Kit (Qiagen). The extracted DNA served as the template in the second PCR primers of PMCF (5’-CTA GTG CGG CCG CTG AGG AGA CGG TGA CCT GGG T-3’) and A6E (5’-GAT GTG CAG CTG CAG GAG TCT GGR GGA GG-3’) were used for the amplification of the VHHs.The purification of PCR products was finalized by QIAquick PCR Purification Kit. Restriction endonucleases ofPstI andNotI were utilized to digest the VHH fragments and phagemid of pMECS. The recombinant phagemid containing the VHH insertion was constructed after overnight incubation with T4 ligase at 16 °C, and then electrotransformed intoE. coliTG1 competent cells (Lucigen) and plated on Luria-Bertani (LB) agar supplemented with 20% (m/V) glucose and 100 µg/mL ampicillin. The Nb library was constructed after collecting all of the colonies on the LB agar plates and stocked with the presence of 20% of glycerol. The diversity of the library was determined after series-dilution and plating on LB agar. The percentage of VHH insertion was assessed by colony PCR with primers of MP57 (5’-TTA TGC TTC CGG CTC GTA TG-3’) and GIII (5’-CCA CAG ACA GCC CTC ATA G-3’) and agarose-gel electrophoresis to visualize the bands corresponding to VHHs.
2.4 Biopanning
Four rounds of bio-panning were performed according to the established protocol for the enrichment of antigen specific Nbs [35].Generally, an aliquot of the library (~1 mL) was added to 300 mL 2 × TY medium supplemented with glucose and ampicillin, and incubated with shaking at 37 °C until the exponential stage reached.The helper phage M13K07 (~1012) was added to the culture, and incubated at room temperature to allow the infection ofE. coli. After overnight incubation, phage particles with fused-expressed Nbs were collected from the supernatant after centrifugation to remove the TG1 cells, and then precipitated with polyethylene glycol (PEG)/NaCl to facilitate the final resuspension in 1 mL sterile Phosphate Buffered Saline (PBS). Microtiter plates (Corning) were coated with 100 µL of extracted antigen at a concentration of 50 µg/mL at 4 °C overnight,and the negative control without coated antigen was used for the base line determination. After blocking of the remaining binding sites with 200 µL of 3% (m/V) skim milk, around 1 × 1011PFU of Nbdisplaying phages were added and incubated at room temperature for 1 h. Unbounded phages were then removed by stringent washing steps with PBST (PBS containing 0.05% Tween-20) for 10 times in 1stpanning round, 20 times in 2ndand 3rdpanning rounds, 10 times in 4thpanning round. The remaining phage particles were eluted with 100 µL of triethylamine (100 mmol/L TEA, pH 11.0) and neutralized with 100 µL Tris-HCl (1.0 mol/L, pH 7.4) immediately after collection. Next, 10 µL of the harvested phages were serially diluted and subsequently added into wells with 90 µL of TG1 cells for enrichment analysis. The remaining phage particles were incubated with TG1 cells to allow the infection and the subsequent amplification of the phages employed for the next round of panning.
2.5 Selection of specific Nbs
For the screening of specific Nbs against peanut extracted proteins,individual colonies were randomly selected after three-rounds of bio-panning and cultured in 100 µL of 2 × TY medium supplemented with 10% (m/V) glycerol, 2% (m/V) glucose and 100 µg/mL ampicillin in 96-well culture plates (Corning). After overnight incubation at 37 °C without shaking, cultured strains were transferred into deep well plates (Axygen) with 2 × TY medium supplemented with 0.1 g/100 mL glucose and 100 µg/mL ampicillin, and cultured with shaking at 37 °C for 3-4 h until the exponential phase was reached. The expression of Nbs was induced by adding isopropylβ-D-1-thiogalactopyranoside (IPTG) to reach a final concentration of 1 mmol/L. Cells were pelleted after centrifugation and applied to several freeze-thaw cycles to release the protein including the Nb. The recognition and binding capability of the released Nbs against coated peanut protein was evaluated through PE-ELISA. Wells coated with PBS instead of the peanut protein were utilized as the negative control to provide background values that should be at least 2-fold below the signal for wells with antigen for the positive binders. The antigen bounded Nbs were detected with mouse anti-hemagglutinin (HA) IgG(Invitrogen) and AP conjugated goat anti-mouse IgG (Invitrogen)as the primary and secondary antibodies respectively. The colonies showed positive signals in PE-ELISA were chosen for the following phagemids extraction and gene sequencing of the inserted VHHs.
2.6 Antigen specificity of Nbs
Western blotting was performed to evaluate the binding specificity of the selected Nbs against peanut protein. Briefly, peanut protein extract was separated by SDS-PAGE, and then transferred to nitrocellulose (NC)membrane (GE Healthcare). The membrane imprinted with peanut protein was cut into paralleled strips after blocking with 3% skim milk before the subsequent incubation with 1 µg/mL of Nbs in sterile PBS at RT separately. After the washing steps with PBST, mouse anti-His tag IgG and horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG were then incubated sequentially as primary antibody and secondary antibody, respectively. The binding of Nbs to antigen was visualized after developing with mixed HRP-substrate staining solutions (solution A: 6 mL methanol with 18 mg chloro-1-naphtol;solution B: 30 mL TPA solution (500 mL: 14.63 g NaCl, 1.4 g Trizma base, pH 7.5) with 19 µL H2O2).
2.7 Expression and purification of selected Nbs
The pMECS plasmids containing the encoding genes of the selected Nbs were extracted and transformed intoE. coliWK6 cells for the production of Nbs with fused HA- and His-tag at the C-terminus. Cells were cultured in Terrific Broth (TB) liquid medium supplemented with 0.1% (m/V) glucose, 100 µg/mL ampicillin and 2 mmol/L MgCl2at 37 °C until an OD600nmof around 0.6 was reached. 1 mmol/L of IPTG was added to induce expression of Nbs in the periplasm, and the expressed protein was released by osmotic shock. Immobilized metal affinity chromatography (IMAC) based on HisPurTMNi-NTA Resin (Thermo Scientific) was performed to purify the His-tagged Nbs. After washing steps, unspecific binding components were removed from the Ni-NTA resin and the tightly bounded His-tagged Nbs was eluted with elution buffer containing 500 mmol/L imidazole. The eluate from the IMAC was applied to size exclusion chromatography (SEC) on a HiLoadTM16/600 SuperdexTM75 pg prepacked column (GE Healthcare) for further purification and buffer exchange. The purity of purified Nbs was determined by SDS-PAGE after Coomassie-blue staining. Western blotting was performed to confirm the identity of the Nbs by using mouse anti-HA monoclonal IgG and HRP conjugated goat anti-mouse IgG. The concentration of Nbs was determined by measuring OD280nmand using the theoretical extinction coefficient of the Nb as calculated from its amino acid content. The purified Nbs were stored at a concentration of 1 mg/mL for further study.
2.8 Targeting analysis by ELISA
Peanut protein at a concentration of 50 µg/mL was coated in micro-well titer plates at 4 °C overnight. After blocking of the residual binding sites with 2% skim milk, 10 µg of purified Nbs were incubated for 1 h at room temperature. Additional set up supplemented with PBS instead of Nbs was employed as blank control. Mouse anti-HA IgG and HRP conjugated goat anti-mouse IgG were used as the primary and secondary antibodies respectively. The absorbance at 450 nm was determined by microplate reader.
2.9 Immunoprecipitation and antigen identification by LC-MS/MS
For the identification of the antigen targeted by the selected Nbs, immunoprecipitation (IP) was performed using HisPurTMNi-NTA magnetic beads (Thermo Scientific) to capture the antigen protein from the peanut extract. Briefly, 250 µL of aliquoted peanut protein extract at a concentration of 1 mg/mL was incubated with 10 µg of Nbs overnight at 4 °C with shaking. Then, 10 µL of Ni-NTA magnetic beads were added into the mixture to capture the Nb-antigen complex by incubation at 4 °C overnight. The magnetic beads were collected using a magnetic stand and washed several times to remove the unspecific binding components. The magnetic beads containing Nb-antigen complex were eluted with 40 µL of elution buffer containing 250 mmol/L imidazole, and then applied to SDS-PAGE and Western blotting to visualize the bands corresponding to the targeted antigen after Coomassie-blue staining and color development respectively. The bands corresponding to the antigen confirmed by SDS-PAGE and Western blotting were excised from the gel and applied for the following preparation process of peptides utilized in LC-MS/MS analysis. Briefly, the excised gel was de-stained completely to remove the residual Coomassie-blue, and then digested with trypsin to produce peptides for analysis with LC-MS/MS (Waters Corporation). For peptide identification, data were generated and analyzed in the Proteome Discoverer 1.4 (Thermo Scientific) with the protein database generated from NCBI and Uniprot. Peptide matches were filtered using theq-value and Posterior Error Probability calculated by the Percolator algorithm ensuring a false detection rate below 5%.
2.10 Characteristics of Nbs
2.10.1 Thermal stability
The thermal stability of the purified Nb was determined by thermofluor in CFX ConnectTMReal-Time PCR System (Bio-Rad).Briefly, the reaction system of 30 µL including 15 µL of Nb, 5 µL of 100 × diluted SYPRO®Orange Protein Gel Stain (Sigma-Aldrich),and 10 µL of PBS was prepared in wells of a micro-titer plate. The system with only SYPRO®Orange Protein Gel Stain and sterile PBS were employed as blank for baseline control. The temperature raising program was set up from 25 °C to 95 °C at a scan rate of 0.5 °C/min to record the fluorescence signal every 0.5 °C. Triplicated samples were prepared to ensure the repeatability. Data analysis was performed with OriginPro 8 software by employing non-linear fitting to determine the melting temperature (Tm) of Nbs.
2.10.2 Affinity and cross-reactivity of Nbs
The affinity and cross-reactivity of Nbs against the antigens were determined by direct ELISA. For the affinity analysis, peanut extract at a concentration of 100 µg/mL was deposited in micro-well titer plates at 4 °C overnight. After blocking of the residual binding sites with 2% skim milk, serially diluted Nbs at a concentration of 10 000, 1 000, 100, 10, 5, 1, 0.5, 0.1, 0.01, 0.001, 0.000 1 nmol/L were added to the wells and incubated for 1 h at room temperature.In addition, set up supplemented with PBS instead of Nbs was employed as negative control for the base line signal. Mouse anti-HA IgG and HRP conjugated goat anti-mouse IgG were used as the primary and secondary antibodies respectively. The absorbance was determined at 450 nm by microplate reader. The apparent affinity of Nbs was defined as the concentration of Nb corresponding to half of maximum signal. For the cross-reactivity of Nbs, protein extract from peanut, macadamia nut and Lupine were coated at a concentration of 100 µg/mL. Nbs were incubated at 100 µg/mL. The incubation of the primary and secondary antibodies, as well as the color development was according to the steps described above.
2.11 Sandwich ELISA
2.11.1 Preparation of Nbs with only His-tag only
Based on the proposal to establish sandwich ELISA with the capturing and detecting antibodies, Nbs with only His-tag were expressed and purified according to the protocol described above.Briefly, the encoding gene fragment was subcloned into pHEN6c plasmid to construct recombinant vector, and then transformed intoE. coliWK6 cells for the production of the cloned Nb in the periplasm. The purification steps of only His-tagged Nbs was consistent with the previous method including IMAC and SEC. The purity and identity of the only His-tagged Nbs were confirmed by SDS-PAGE and Western blotting using anti-His antibodies as probe.
2.11.2 Establishment of sandwich ELISA
For the development of sandwich ELISA, Nbs with only Histag and both tags (His- and HA-tag) were utilized as the capturing and detecting antibodies respectively. Nb-based sandwich ELISA was performed to determine the optimized concentration of the capturing and detecting Nbs through a checkerboard titration with a series of dilutions. Basically, the capturing Nb was coated at various concentrations from 0 to 10 µg/mL. After blocking with 5% skimmed milk, 100 µL of peanut protein at a concentration of 50 µg/mL was added and incubated for 1 h at room temperature. Then, the detecting Nb was incubated at different concentration from 0 to 10 µg/mL.Mouse anti-HA IgG and HRP-conjugated goat anti-mouse IgG were employed as the primary and secondary antibodies respectively.TMB substrate was utilized for the development of signal, and the optical density at 450 nm was recorded by a microplate reader. The concentration of the capturing and detecting Nbs was optimized to develop the most sensitive sandwich ELISA. The calibration curve and linear standard curve was provided with the natural logarithm of the macadamia protein concentration and OD450nmvalue. For the developed sandwich ELISA, the limit of detection (LOD) and the limit of quantitation (LOQ) were determined by testing negative wells with PBS, and calculated as the protein concentration corresponding to the mean of 10 blank values plus 3 or 10 times the standard deviations (SD), respectively.
2.11.3 Analysis of spiked samples
The applicability of the established sandwich ELISA was evaluated against the dairy product of skim milk purchased from the local supermarket. The spiked sample was prepared after supplementing various concentrations of peanut protein extract (defatted, and diluted 1 000 times). The spiked samples were then applied for the sandwich ELISA to determine the recovery rate and coefficient of variation (CV) for the assessment of the precision and reproducibility of the sandwich ELISA.
3. Results and discussion
3.1 Preparation of Peanut protein extracts
A crude extract of peanut proteins was successfully prepared following the well-established protocol and yielded an acceptable recovery of more than 90%. The complexity of the extracted proteins was revealed by Coomassie-blue stained SDS-PAGE (Fig. 1)indicated the distribution of molecular weight mainly ranging from 10 to 70 kDa, which is in agreement with previous results [36]. The significant difference in the position of protein distribution under reducing and non-reducing conditions indicated the high content of natural polypeptides with disulfide bonds in the peanut protein extract.The concentration of peanut protein was determined using the BCA detection kit to be around 34 mg/mL for our stock.

Fig. 1 SDS-PAGE analysis of peanut protein extract. The protein extract was separated by utilizing 12% gel under reducing (R) and non-reducing (NR)conditions respectively.
3.2 Construction of Nb library
A young alpaca was immunized with peanut protein extract to raise an immune response.VHHgenes corresponding to the size around 400 bp were obtained after a two-step nested PCR. After the first round PCR, two fragments corresponding to the size of 900 and 700 bp were visualized reflecting the amplified products corresponding to VH-CH1-CH2 regions and VHH-CH2 regions respectively. The second PCR with the extracted 700 bp fragment as the template yielded the VHH fragments corresponding to the size of around 400 bp. After the construction of the Nb library, the size was determined after serial dilutions and colony counting to be 1.44 × 107CFU/mL. Furthermore, the percentage of clones with a correctly sized insert into the phage display vector was assessed by colony PCR of 40 randomly picked single colonies and shown to approach 75% (data not shown).
3.3 Bio-panning and screening of Nbs
Bio-panning was performed to enrich the potential binders against the peanut protein extract. The efficient enrichment of the consecutive bio-panning rounds was verified by comparing the number of colonies originated from the well of micro-titer plates coated with or without antigen, and the enrichment was noted to increase from 1.2 to 7.3-fold in consecutive rounds of bio-panning (Fig. 2A).
In order to further screen the specific Nbs, 190 single colonies were randomly selected from four rounds of panning (24 single colonies from 1stround, 48 colonies from 2ndround, 59 colonies from 3rdround, and 59 colonies from 4thround), and applied for the following PE-ELISA for the specific Nb selection. The results revealed 34 colonies yielding a high signal when compared to the absorbance from the control wells without antigen (Fig. 2B). The selected positive binders were all included for gene sequencing to identify the Nb families. The results indicated the retrieval of 6 Nbs belonging to different families according to the amino acid within the third antigen binding loop and referred to as P43, P84, P99, P72,P91 and P178. The Nb sequences were aligned according to the‘International Immunogenetics Information System (IMGT)’ and are shown in Fig. 2C.
3.4 Specificity of selected Nbs
After selection of specific Nbs, the specificity was first assessed based on the periplasm extraction containing the specific Nbs from theE. coliWK6 cells via Western blotting. The extracted peanut proteins were separated by the SDS-PAGE and then transferred to the NC membrane. The results indicated the targeting of Nb P43 to the proteins with a corresponding size of around 60 kDa (Fig. 3). The presence of two distinctive bands of slightly different molecular mass,targeted by P43 in Western blotting could indicate a high degree of sequence identity within these antigens. However, this needs to be confirmed in following investigations. No bands were visualized when using the other Nbs as probe in Western blotting, which most likely indicated the targeting of these Nbs to conformational epitopes on the peanut antigens.

Fig. 2 Bio-panning and screening of specific Nbs. (A) Evaluation of the enrichment efficiency. (B) 34 positive binders screened by PE-ELISA with green bars for the signal in wells with antigen coated and gray bars for response in wells without antigen. (C) Amino acid sequence alignment of the selected Nbs.

Fig. 3 The specificity analysis of selected Nbs was determined by Western blotting. Lane B, the group using the other Nbs as probe.
3.5 Expression and purification of specific Nb
Nb P43 was purified according to the standard procedure.Plasmids containing the encoding gene of P43 was transformed intoE. coliWK6 cells for the periplasmic production after induction with IPTG. Osmotic shock was performed to prepare the soluble extraction,and the purified formats were obtained through a two-step purification protocol encompassing IMAC and SEC (Fig. 4A). The relatively high yield was determined to be around 10 mg/L bacterial culture in TB medium. The purity of prepared Nb was analyzed by SDS-PAGE to reveal the presence of a single band with molecular weight round 15 kDa as expected according to their amino acid sequences (Fig. 4B).Western blotting was performed to verify the identity of Nbs with the presence of His-tag (Fig. 4C).
3.6 Identification of Nb-targeted antigen by LC-MS/MS
The target allergen of Nb P43 was immuno-captured by utilizing HisPurTMNi-NTA magnetic beads. The formed Nb-antigen complex was separated on SDS-PAGE, and revealed the proteins with a molecular mass mainly ranging from 50 to 70 kDa (Fig. 5A). Based on the signal of the Western blotting (Fig. 4, 5B), two bands with lower size were extracted for the identification by LC-MS/MS, and the results revealed the identity of Ara h 3 (UniProtKB: E5G077) and Gly 1(UniProtKB: Q9FZ11) as corresponding to the identified peptides in Fig. 5C and 5D. In order to explain the cross-reaction of Nb P43 on Ara h 3 and Gly 1, the amino acid similarity among these proteins was investigated. The detailed analysis demonstrated that Gly 1 was belonging to the Ara h 3 family. The alignment of Gly 1 and Ara h 3 revealed a high degree of sequence identity (72%), which provided a reasonable explanation for the cross reactivity of Nb P43 (Fig. 6). The targeting of Nb P43 to both Ara h 3 and Gly 1 could be considered beneficial for the development of an immuno-assay for the efficient surveillance of peanut contamination in food matrix.
3.7 Characteristics of Nb
3.7.1 Thermal stability
The thermal stability of the Nb P43 was monitored by thermofluor in a real-time PCR detection system. The Nb P43 with aTmof(77.34 ± 0.09) °C was demonstrated to possess a high thermal stability (Fig. 7A).
3.7.2 Affinity and cross-reactivity
The apparent affinity of Nb P43 against peanut protein extract was estimated by direct ELISA to be around 1 nmol/L (Fig. 7B), which reflected the high affinity of Nb P43. The cross-reactivity of Nb P43 was determined by performing an ELISA against proteins extracted from macadamia and lupine. These results demonstrated the relatively low cross-reactivity among proteins extracted from various nuts (Fig. 7C).
3.8 Establishment of sandwich ELISA
3.8.1 A pair of Nb P43 can capture and detect the antigen in a sandwich ELISA
The use of Nb P43 with an His-tag as antigen capturing antibody and Nb P43 with His- and HA-tag for detecting the captured antigen seems to work in a sandwich ELISA to monitor the presence of peanut proteins. As shown in Fig. 8A, the high signal response obtained in ELISA demonstrated the application of the same Nb P43 to clamp the antigen, and provided the experimental evidence for an easy development of an immuno-assay based on a pair of Nb P43 to detect the presence of peanut allergen. The optimal concentration of capturing and detecting antibodies was determined. As verified by the results from sandwich ELISA, where various concentrations of capturing and detecting Nbs were tested, the Nb concentration corresponding to high signal and low background was selected for the development of the sandwich, with the optimal concentrations of 5 µg/mL and 7.5 µg/mL for the capturing and detecting antibodies respectively (Fig. 8B).

Fig. 4 Purification and confirmation of Nb P43. (A) Purification of P43 by SEC with the Nb fraction. (B) SDS-PAGE and (C) Western blotting of purified Nb P43.

Fig. 5 Identification of antigen protein targeted by Nb P43. (A) The captured proteins after immunoprecipitation were separated and visualized by SDS-PAGE.Lane B, the control group without supplemented Nb served as the blank. The bands besides the indicated Ara h 3 and Gly 1 could potentially represent the peptides or precursors originating from Ara h 3 and Gly 1. (B) Western blotting was performed to visualized the bands identified by Nb P43. (C) The identified peptides corresponding to the amino acid sequence of Ara h 3 were indicated in purple boxes. (D) The identified peptides corresponding to the amino acid sequence of Gly 1 were indicated in green boxes.

Fig. 6 The amino acid sequence alignment between Ara h 3 and Gly 1. The homologous alignment was highlighted with the yellow background to indicate the high level of similarity between Ara h 3 and Gly 1.
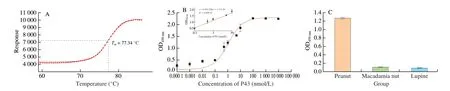
Fig. 7 Characteristics of Nb P43. (A) Thermal stability of Nb P43. (B) Affinity of Nb P43. (C) The cross reaction of Nb P43 with protein extract from macadamia nut and lupine.

Fig. 8 Applicability of self-paired Nb and the optimal concentration of Nb-pair for the development of sandwich ELISA. (A) The corresponding absorbance value of sandwich ELISA based on self-paired Nb P43. N-HA is the control group without capturing antibodies, N-His represented the control without detecting antibodies. (B) The optimal concentration of capturing and detecting antibodies.
3.8.2 Standard curve of developed sandwich ELISA
After a relevant optimization process, the sandwich ELISA was developed for the detection of peanut allergen. A calibration curve was obtained with a linear range from 0.2 to 10.6 µg/mL. The fitted equation of the calibration curve was determined asy= 1.159 67lnx+0.877 52, with the correlation coefficient of 0.992 22 (R2,n= 3).The calculated LOD and LOQ were 53.13 ng/mL and 189.11 ng/mL respectively for the crude peanut protein extract (Fig. 9).

Fig. 9 The standard curve of the developed sandwich ELISA. The embedded graph represented the linear range of the immuno-assay. The linear equation was calculated as y = 1.159 67lnx + 0.877 52 with an acceptable correlation coefficient (R2) of 0.992 22.
3.8.3 Analysis of the spiked food sample
The effectiveness of the developed sandwich ELISA was tested for crude peanut protein extract spiked in milk at different concentrations. Each spiked sample was tested in triplicate to ensure the accuracy of the determination. As shown in Table 1, the recovery was demonstrated to be between 93.8% and 114.7%, which provided significant evidence for the credibility of the established Nb-based sandwich ELISA to detect peanut allergen in food matrix.Interestingly, the selected Nb P43 is applicable for the surveillance of residual peanut components by detecting the allergen components of Ara h 3 and Gly 1.
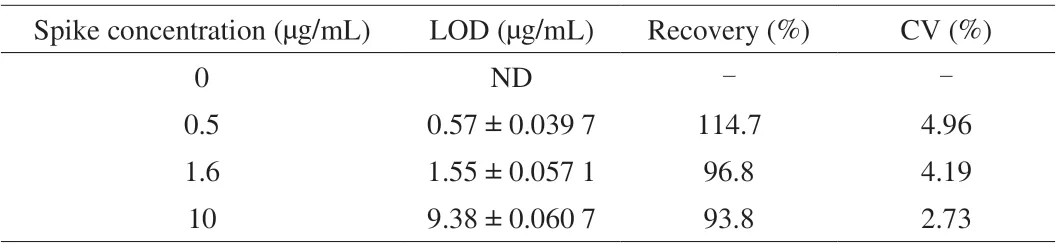
Table 1 Analysis of the skim milk sample spiked with different concentration of peanut protein extract (n = 3).
4. Discussion
The current study reported the selection of specific Nbs against the crude peanut protein extract by using a total protein extract without enrichment steps as immunogen in alpaca and also for subsequent selection of monoclonal Nbs. The addressed strategy could facilitate the identification of specific Nbs against peanut allergen without the necessity to prepare recombinant allergen as usual. The retrieved 6 Nbs confirmed the validity of this strategy for the identification of binders against protein components, especially the Nb P43 against peanut allergen of Ara h 3. Only Nb P43 has been investigated to monitor the presence of the target antigen, as it is the only Nb that could be used in Western blotting and for which we could determine the antigen.
The cross reaction of Nb P43 against Ara h 3 and Gly 1 is a favorable property to establish a sandwich ELISA, as the surveillance of peanut contamination with high sensitivity could be expected. The application of the developed sandwich ELISA on the detection in food matrix will not only yield the response from peanut allergen of Ara h 3,but also the signal from Gly 1 to amplify the detection response, and facilitate the sensitive surveillance of the peanut contamination in food samples. Interestingly, the established sandwich ELISA could be applied to quantify the presence and contamination of peanut protein components. Generally, it’s preferred to employ different Nbs as the capturing and detecting antibodies for the development of sandwich ELISA, as it could avoid the restricted detection signals resulting from the epitope binding competition. Initially, it has not been taken into the consideration to establish the sandwich ELISA based on the self-paired Nb. The proceeded research interests were depending on the polymeric formats of the peanut allergen of Ara h 3 and Gly 1,which could facilitate the multiple Nb binding epitopes to fulfill the interaction with capturing and detecting Nbs simultaneously. The integration of the selected Nbs with other detection techniques, such as fluorescent or electrochemistry-based inspection could further enhance the detection of peanut allergen with raised sensitivity and effectiveness.
The current research on the design of sandwich ELISA for peanut allergen reveals a general strategy to identify target specific Nbs that could facilitate the further development of alternative,simple and sensitive immune detection strategies for allergen surveillance.
5. Conclusions
The current study reported the successful identification of Nbs against peanut allergen protein, and provided evidence for the application of non-enriched immunogen and antigens during selection. The detection strategy of the sandwich ELISA against peanut allergen was verified with the selected Nb for the surveillance of peanut protein contamination in food matrix. The high affinity and sensitivity were demonstrated for the established immuno-assay, as well as the potential applicability in real food samples. Furthermore,the combination of the selected Nbs with more advanced detection techniques can facilitate the establishment of a more practical immuno-assay with higher sensitivity.
Declaration of competing interest
The authors declare that they have no competing financial interests.
Acknowledgements
This study was financially supported by the grants from the National Key R & D Program of China (No. 2019YFC1605005).

- 食品科学与人类健康(英文)的其它文章
- The role of probiotics in prevention and treatment of food allergy
- Roles of fermented plant-, dairy- and meat-based foods in the modulation of allergic responses
- The role of gut microbiota and its metabolites short-chain fatty acids in food allergy
- Association of nutrients intake during pregnancy with the risk of allergic disease in offspring: a meta-analysis of prospective cohort studies
- Purif ication and immunoglobulin E epitopes identif ication of low molecular weight glutenin: an allergen in Chinese wheat
- Determination of egg and milk allergen in food products by liquid chromatography-tandem mass spectrometry based on signature peptides and isotope-labeled internal standard