
白果仁及炒白果仁内源性有害成分分析
2023-06-04 07:00张雁淼丁明和谈仲川周天天干国平1b
湖北农业科学 2023年5期
张雁淼,丁明和,张 营,谈仲川,周天天,干国平,1b
(1.湖北中医药大学,a.药学院;b.湖北省中药炮制工程技术研究中心,武汉 430065;2.马应龙药业股份有限公司,武汉 430064)
白果为银杏科植物银杏(Ginkgo bilobaL.)的干燥成熟种子,药食同源[1]。中国白果资源丰富,年产量约2 万t[2]。白果中含有银杏叶黄酮内酯[3]、多元醇等活性成分,并含有丰富的多糖[3,4]、蛋白质、淀粉、脂肪、维生素C、氨基酸和矿物质等营养成分[4],具有良好的临床疗效和食用价值[5-7]。但长期以来,由于白果有一定毒性,严重制约了白果资源的开发利用[8]。现代研究表明,白果中内源性有害成分为总银杏酸、吡哆醇、氰苷。总银杏酸为水杨酸的6-烷基或6-烯基衍生物,主要由银杏新酸、银杏酸、氢化银杏酸、氢化银杏酸、银杏二酚等组成,具有细胞毒性、神经毒性、免疫毒性[9],含量过高会导致腹痛、腹泻、过敏性皮炎等不良反应[10]。吡哆醇主要由4′-O-甲基吡哆醇(MPN)、4′-O-甲氧基吡哆醇-5′-葡萄糖苷(MPNG)等组成[11],统称为银杏毒(TMPN),含量过高会引起呕吐、腹泻、抽搐癫痫等[12]。氰苷成分通过体内代谢分解为氢氰酸,氢氰酸可导致细胞缺氧死亡引起呼吸加快加深、心律不齐、脉搏加快、抽搐昏迷,最后意识丧失,呼吸衰竭而亡[13]。
白果生食有毒,中医认为炒制可降低毒性[14],但未见炒制白果可降低毒性的关键证据。虽为药食同源品种,却有食用白果和食用炒白果中毒的报道[15]。迄今为止,未见对白果及其饮片内源性有害成分的分析研究,《中国药典》中亦未对白果仁及其炮制品炒白果仁中内源性有害成分进行含量控制和安全性评价,因此无法保证白果及其饮片的安全性。本研究采用HPLC 法、HS-GC 法分别建立白果仁中总银杏酸、吡哆醇、氰苷的含量分析方法,为白果及其饮片的质量控制、安全用药和白果的开发利用提供科学依据。
1 材料与方法
1.1 材料与试剂
10 批白果药材分别采自湖北省安陆市、湖北省巴东县、湖北省武汉市黄陂区、江苏省徐州市邳州市、江苏省泰兴市新街镇、河南省信阳市新县、山东省临沂市郯城县、重庆市涪陵区、云南省保山市腾冲市、广西壮族自治区桂林市灵川县(样品编号依次为1 号至10 号)。经湖北中医药大学湖北省中药炮制工程技术研究中心鉴定为银杏科植物银杏的种子。标本保存于湖北中医药大学药物化学研究室1 室。
供试品白果仁(生品)处理:白果去壳,参照《中国药典》[16]掸法(通则0213)去内种皮;供试品炒白果仁(炒制品)处理:参照《中国药典》[16]清炒法(通则0213)制备,白果仁、炒白果仁均经粉碎(过3 号筛)处理。
白果新酸对照品、总银杏酸对照品,成都埃法生物科技有限公司;MPN 对照品,成都普思生物科技股份有限公司;MPNG 对照品,自制,HPLC 归一化法测定纯度为98.7%;水中氰成分分析标准物质,GBW(E)080115,中国计量科学研究;甲醇,色谱纯,常熟市鸿盛精细化工有限公司;乙腈,色谱纯,西格玛奥德里奇(上海)贸易公司;氯胺T,分析纯,国药集团化学试剂有限公司;磷酸、盐酸、氢氧化钠、甲基橙、三氟乙酸均为分析纯。
1.2 仪器与设备
LC-20AD 型高效液相色谱仪(配有SPD-20A 型紫外可见光检测器及LabSolutions 数据处理软件),日本岛津公司;Aglient7890B 型气相色谱仪(配有电子捕获检测器)、Aglient7697A 型顶空进样器,美国安捷伦科技有限公司;AS 系列超声波清洗机,天津奥特赛恩斯仪器有限公司;BS210S 型十万分之一天平,北京赛多利斯天平有限公司;hhS-2S 型电子恒温水浴锅,上海康华生化仪器制造有限公司。
1.3 色谱条件
1)总银杏酸色谱条件。
色谱柱为YMC-Triart C18(4.6 mm×150 mm,5 μm),以0.1%三氟乙酸的乙腈溶液为流动相A,以0.1%三氟乙酸的水溶液为流动相B,进行梯度洗脱(表1)。柱温为30 ℃,检测波长为310 nm,流速为1.0 mL/min,进样量为20 μL。

表1 总银杏酸流动相洗脱程序
2)MPN 和MPNG 色谱条件。
色谱柱为YMC -Triart C18(4.6 mm×150 mm,5 μm);以甲醇为流动相A,以去离子水为流动相B,进行梯度洗脱(表2)。柱温为30 ℃,检测波长为328 nm,流速为1.0 mL/min,进样量为10 μL。

表2 MPN 和MPNG 流动相洗脱程序
3)氢氰酸色谱条件。
色谱柱为DB-WAX 毛细管柱(30 m×0.25 mm,0.25 μm);色谱柱温度:40 ℃保持5 min,以50 ℃/min速率升至200 ℃,保持2 min;载气:N2;进样口温度:200 ℃;检测器温度:250 ℃;尾吹:60 mL/min;分流比:5∶1;柱流速:2.0 mL/min。顶空平衡温度:50 ℃;平衡时间:30 min。
1.4 溶液的制备
1)总银杏酸。
对照品溶液:精密称取白果新酸对照品2.03 mg,置于50 mL 容量瓶中,加甲醇溶解并稀释至刻度,摇匀(浓度为40.60 μg/mL)。
定位用对照品溶液:精密称取总银杏酸对照品1.08 mg,置于10 mL 容量瓶中,加甲醇溶解并稀释至刻度,摇匀,即得(浓度为108.00 μg/mL)。
供试品溶液:取样品粉末4.00 g,精密称定,置于具塞锥形瓶中,精密加入甲醇20 mL,称定重量,水浴回流3 h,放冷至室温,用甲醇补足减失的重量,摇匀,用微孔滤膜过滤,取续滤液,即得。
2)MPN 和MPNG。
对照品溶液:精密称取MPN 对照品4.95 mg,置于50 mL 容量瓶中,加入甲醇溶解并稀释至刻度,摇匀(浓度为99.00 μg/mL)。精密称取MPNG 对照品5.60 mg,置于50 mL 容量瓶中,加入甲醇溶解并稀释至刻度,摇匀(浓度为112.00 μg/mL)。
供试品溶液:取样品粉末1.00 g,精密称定,置于具塞锥形瓶中,精密加入去离子水50 mL,称定重量,超声30 min,放冷,用去离子水补足减失的重量,离心10 min,用微孔滤膜过滤,取续滤液,即得。
3)氢氰酸。
对照品溶液:精密吸取水中氰成分分析标准物质2 mL,置10 mL 容量瓶中,用0.1%氢氧化钠溶液定容(浓度为10 mg/L),用去离子水稀释,配制浓度为0、0.001、0.002、0.010、0.050、0.100 mg/L 的氰离子(以CN-计)标准工作溶液。
供试品溶液:取样品粉末约1.00 g,精密称定,置100 mL 容量瓶中,加去离子水适量,超声30 min,放冷,定容至刻度,4 000 r/min 离心5 min,精密吸取上清液10 mL,置顶空瓶中,加16.7% 磷酸溶液0.2 mL,涡旋混合,加氯胺T 溶液0.2 mL,立即加盖密封,涡旋混合,即得。
1.5 定量限与检测限
精密吸取对照品溶液,用甲醇逐级稀释至信噪比S/N=10 时即为定量限,至信噪比S/N=3 时即为检测限。
1.6 标准曲线绘制
1)总银杏酸标准曲线。
分别精密吸取白果新酸对照品溶液(浓度为40.60 μg/mL)1、2、3、4、5 mL,加入甲醇定容至10 mL容量瓶,混匀,制成一系列不同浓度的溶液,进样分析。以峰面积(y)对溶液浓度(x)进行线性回归。
2)MPN 和MPNG 标准曲线。
分别精密吸取MPN 对照品溶液(浓度为99.00 μg/mL)120、160、200、240、280 μL,加入甲醇定容至10 mL,混匀,制成一系列不同浓度的溶液,进样分析。精密吸取MPNG 对照品溶液(浓度为112.00 μg/mL)0.6、0.8、1、1.2、1.4 mL,加入甲醇定容至10 mL,混匀,制成一系列不同浓度的溶液,进样分析。以峰面积(y)对溶液浓度(x)进行线性回归。
3)氢氰酸标准曲线。
分别取0、0.001、0.002、0.010、0.050、0.100 mg/L氰离子标准工作溶液各10.0 mL,置于6 个顶空瓶中,加入0.2 mL 磷酸溶液,涡旋混合,然后加入0.2 mL 氯胺T 溶液,立即加盖密封,涡旋混合,进样分析。以峰面积(y)对溶液浓度(x)进行线性回归。
1.7 精密度试验
按“1.4”项方法制备6 份供试品溶液,进样分析,测定其峰面积,计算精密度。
1.8 溶液的稳定性试验
分别取同浓度的供试品溶液和同浓度的对照品溶液,置于室温下,于0、2、4、6、8 h 分别进样分析。
1.9 回收率试验
1)总银杏酸。
精密称取样品6 份,每份2.00 g,置于具塞锥形瓶中,精密称取白果新酸对照品1.827 mg,置于200 mL 容量瓶中,加甲醇溶解并稀释至刻度,即得白果新酸对照品溶液(9.135 μg/mL),加入上述溶液20 mL,添加量分别为含药量的50%,按“1.4”项下供试品溶液处理方法制备,进样分析,计算回收率。
2)MPN 和MPNG。
精密称取样品6 份,每份0.50 g,置于具塞锥形瓶中,分别精确吸取MPN 对照品溶液(浓度为99.00 μg/mL)、MPNG对照品溶液(浓度为112.00 μg/mL)各4、8 mL,置于200 mL 容量瓶中,加甲醇溶解并稀释至刻度,摇匀,即得MPN 对照品溶液(浓度1.98 μg/mL)、MPNG 对照品溶液(浓度4.48 μg/mL),加入上述溶液各25 mL,添加量分别为含药量的50%,按“1.4”项下供试品溶液处理方法制备,进样分析,计算回收率。
2 结果与分析
2.1 总银杏酸的测定
2.1.1 系统适用性与专属性 精密吸取总银杏酸的对照品溶液、定位用对照品溶液、供试品溶液各20 μL,进样分析。结果(图1)表明,总银杏酸供试品溶液色谱中可检出除白果新酸外的4 种银杏酸成分,且4 种银杏酸成分峰与其他成分峰之间的分离度均大于1.5。

图1 白果中总银杏酸色谱
2.1.2 定量限与检测限 按照“1.5”项方法测定,总银杏酸的定量限为12.18 ng,检测限为4.87 ng。
2.1.3 标准曲线绘制 按照“1.6”项方法测定,总银杏酸回归方程为y=10 218x+836.28,r=0.999 3,表明在浓度为0.609~20.30 μg/mL 时线性关系良好。
2.1.4 精密度试验 按照“1.7”项方法测定,6 份供试品溶液总银杏酸平均含量为90.08 μg/g,RSD=5.12%,表明该方法精密度良好。
2.1.5 溶液的稳定性试验 按照“1.8”项方法测定,供试品溶液总银杏酸峰面积的RSD为2.66%、对照品溶液白果新酸峰面积的RSD为3.79%,表明供试品溶液和对照品溶液在8 h 内稳定。
2.1.6 回收率试验 按照“1.9”项方法测定,总银杏酸的平均回收率为101.17%,RSD为4.57%(n=6)(表3),表明该方法测定结果准确、可靠。

表3 总银杏酸的回收率试验结果
2.2 MPN 和MPNG 的测定
2.2.1 系统适用性与专属性 精密称取MPN 对照品溶液、MPNG 对照品溶液、定位用对照品溶液、供试品溶液各10 μL,进样分析。结果(图2)表明,供试品溶液色谱中,MPN 峰、MPNG 峰与其他成分峰之间的分离度均大于1.5。

图2 白果中MPN、MPNG 色谱
2.2.2 定量限与检测限 按照“1.5”项方法测定,MPN 的定量限为5.94 ng,检测限为2.38 ng,MPNG的定量限为11.20 ng,检测限为4.48 ng。
2.2.3 标准曲线绘制 按照“1.6”项方法测定,MPN的回归方程为y=17 070x+22 618,r=0.999 8,表明在浓度为1.188~2.772 μg/mL 时线性关系良好;MPNG的回归方程为y=5 444.7x-202.79,r=0.999 8,表明在浓度为1.12~15.68 μg/mL 时线性关系良好。
2.2.4 精密度试验 按照“1.7”项方法测定,6 份供试品溶液中MPN 的平均含量为110.36 μg/g,RSD=2.21%;MPNG 的平均含量为242.19 μg/g,RSD=2.57%,表明该方法精密度良好。
2.2.5 溶液的稳定性试验 按照“1.8”项方法测定,供试品溶液MPN、MPNG 峰面积的RSD分别为0.50%、1.87%,对照品溶液MPN、MPNG 峰面积的RSD分别为0.40%、1.61%,表明供试品溶液和对照品溶液在8 h 内稳定。
2.2.6 回收率试验 按照“1.9”项方法测定,MPN、MPNG 的平均回收率为103.14%、103.54%,RSD分别为4.80%、3.05%(n=6)(表4),表明该方法测定结果准确、可靠。
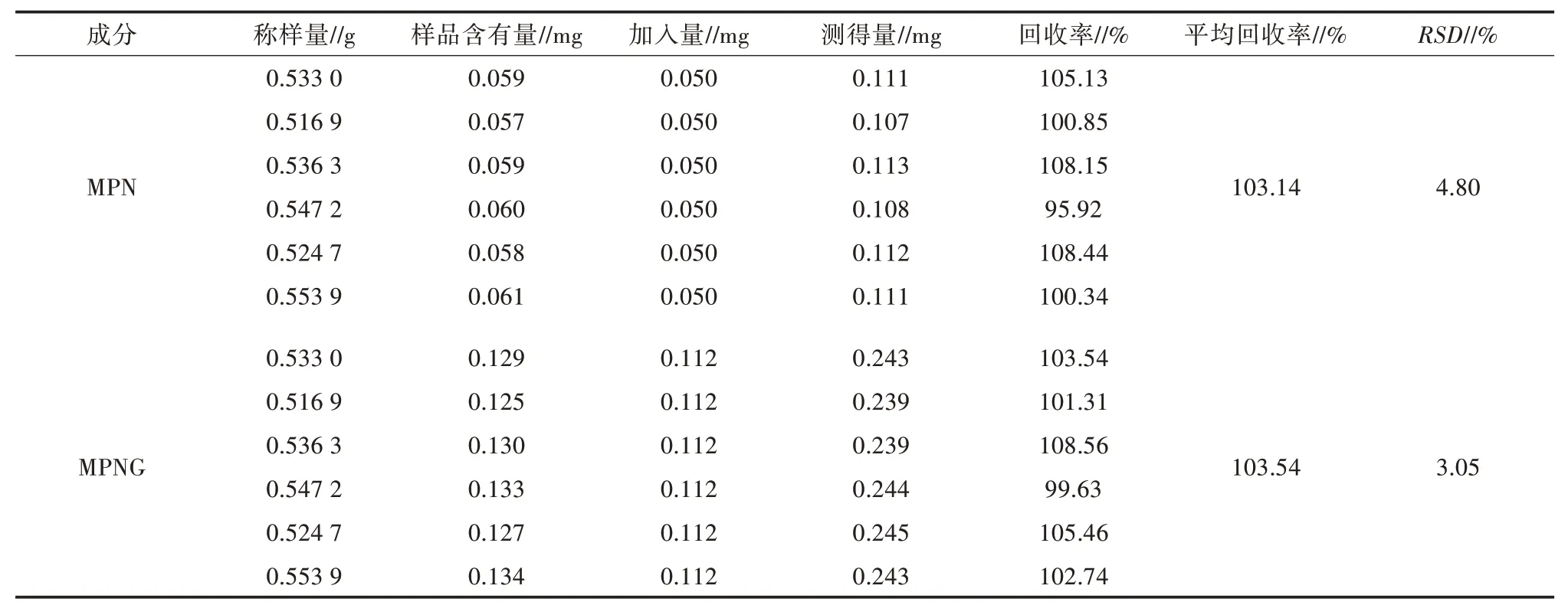
表4 MPN、MPNG 的回收率试验结果
2.3 氢氰酸的测定
2.3.1 系统适用性与专属性 顶空进样分析氢氰酸的空白溶液(浓度为0 mg/L)、对照品溶液(浓度为0.001 mg/L)、供试品溶液。结果(图3)表明,氯化氰与其他成分峰之间的分离度大于1.5,空白溶液无干扰。

图3 白果中氰苷含量测定色谱
2.3.2 样品提取时间和衍生化酸度的选择 以去离子水为溶剂,样品分别采用不同时间(20、30、40 min)进行超声波提取。结果表明,超声波处理30 min 时被测成分可提取完全。
氰化物衍生化需在弱酸性条件下进行,因此分别向顶空瓶中加入100、200、300、400 μL 磷酸溶液来考察酸度条件对衍生化反应的影响。结果表明,加入16.7%磷酸溶液300 μL 时,衍生化反应完全,被测成分峰面积达到最大值。
2.3.3 平衡温度和平衡时间的选择 分别采用不同平衡温度(45、50、55 ℃),不同平衡时间(20、30、40 min),结果表明,当平衡温度为50 ℃、平衡时间为30 min 时被测成分峰面积达到最大值。
2.3.4 定量限与检出限 按照“1.5”项方法测定,氢氰酸定量限为10 ng,检测限为3.5 ng。
2.3.5 标准曲线绘制 按照“1.6”项方法测定,氢氰酸的线性方程为y=9 770.7x+6.062 3,r=0.999 8,n=6,表明浓度为0~0.1 μg/mL 时线性关系良好。
2.4 不同产地的白果仁、炒白果仁中内源性有害成分分析
总银杏酸成分存在于银杏叶及其果实中。《中国药典》对银杏叶中总银杏酸作了限度要求(总银杏酸不得超过10 μg/g),但对白果及其炮制品中总银杏酸未进行限度规定。由表5 可知,10 批不同产地的白果仁中总银杏酸含量为48.597~229.822 μg/g,炒白果仁中总银杏酸含量为44.973~151.058 μg/g。虽然经炮制处理后的各样品中总银杏酸含量与白果仁比较,均有所降低,但残留量还是超过了银杏叶中总银杏酸的限度要求。

表5 不同产地的白果仁、炒白果仁中内源性有害成分的含量(单位:μg/g)
10 批不同产地白果仁中MPN 的含量为19.363~124.782 μg/g,MPNG的含量为265.896~814.585 μg/g,MPNG 含量远高于MPN。炒白果仁中MPN 的含量为18.773~125.505 μg/g,与白果仁比较,7 个样品的MPN 含量均减少,3 个样品(2 号、3 号、5 号)的MPN含量升高;MPNG 的含量为24.646~687.892 μg/g,MPNG 含量均有所减少。但炒白果仁中MPN 和MPNG 的总含量仍有102.729~688.850 μg/g,表明白果通过传统的炮制方法难以达到明显的减毒效果。
欧盟第88/388/EEC 号指令规定灌装果核饮料中氢氰酸限度为5 mg/kg。10 批不同产地白果仁中氢氰酸含量为0~0.176 μg/g,远低于欧盟标准,说明白果中氰苷成分不是主要内源性有害成分。
2.5 不同产地、不同炮制方法白果仁中内源性有害成分的主成分分析
本研究使用Origin 软件对10 批不同产地白果仁、炒白果仁中内源性有害成分的含量进行主成分分析(Principal Component Analysis,PCA)(图4)。特征值大于1 的主成分(PC)分别为PC1(46.3%)、PC2(25.7%)和PC3(21.2%),由于白果仁中MPNG 的含量最高,所以白果仁的位置位于图4a 的右上方,炒白果仁的位置位于下方,可能是由于处理后总银杏酸和MPNG 的含量均有所降低。炒制处理后较高的MPN 保留量决定了样品1 号、2 号和3 号的位置。从PCA 分析看出,白果仁和炒白果仁具有差异性。图4b 中样品4 号、5 号、6 号、7 号具有相近的MPNG 含量,样品8 号、9 号具有相近的总银杏酸含量。由于样品9 号总银杏酸含量最高,所以位于图4b 的最上方,样品1 号MPN 和氰苷含量最高,所以位于图4b的左上方。总之,PCA 分析结果表明不同产地之间白果仁内源性有害成分的含量具有一定的相似性和差异性。

图4 PCA 分析的载荷和得分
3 小结与讨论
氰苷成分在人体中可代谢为氢氰酸,花彤彤等[17]认为白果中氰苷成分为主要内源性有害成分。氰化物的测定方法有分光光度法、气相色谱法和苦味酸试纸法。苦味酸试纸法灵敏度低,分光光度法本身没有分离功能且常受其他成分干扰,气相色谱法分离能力强、检测灵敏度高;倪洋[18]采用酸水解异烟酸-巴比妥酸法在全白果和白果胚乳中检测到微量的氰化物;杨剑婷等[19]采用普鲁士蓝法、苦味酸试纸法和硝酸银滴定法对白果胚和胚乳进行氰化物的测定,均未检测到氰化物;本研究采用灵敏度高的顶空气相色谱法测定。MPN 和MPNG 均具有神经毒性,是白果的主要有害成分。Kobayashi 等[20]、Chang 等[21]认为白果中主要的有害成分为MPN,炒白果仁中主要的有害成分为MPNG,MPNG 毒性小于MPN 的毒性,通过炮制可以降低白果毒性;单舒筠[22]报道白果炮制后MPN 含量降低,与本研究结果一致。
白果虽为药食同源品种,但不同产地白果仁中内源性有害成分(吡哆醇和总银杏酸)含量高、差别大,传统炮制方法制得的炒白果仁中仍含有大量的内源性有害成分,临床应用和食用均存在严重的安全隐患,建议国家主管部门重新审视白果的药食同源属性,同时根据白果内源性有害成分的性质及其毒性作用机理,加强采用现代炮制技术对白果进行炮制减毒的研究,在《中国药典》白果及其饮片质量标准中增加内源性有害成分的检查项目并作限度规定。
本研究建立了有效检测白果仁、炒白果仁中内源性有害成分的方法,并对方法的可靠性性和检出能力进行评价。结果表明,该方法专属性强、重复性好,可有效进行质量控制,为白果的食用和临床使用提供依据。
猜你喜欢
昆明医科大学学报(2021年8期)2021-08-13
幼儿教育·父母孩子版(2021年6期)2021-08-05
科学(2020年3期)2020-11-26
学生天地(2020年5期)2020-08-25
学生天地(2020年3期)2020-08-25
少年漫画(艺术创想)(2020年9期)2020-03-19
幼儿教育·父母孩子版(2020年8期)2020-03-04
基层中医药(2018年11期)2019-01-31
学生天地(2017年7期)2017-05-17
饮食科学(2016年3期)2016-07-04
