
海洋链霉菌MA12的次级代谢产物及抗真菌活性研究
2023-09-06 16:13李宏基张梦雪张天勇樊少武周金华吴文惠
海洋渔业 2023年3期
陈 硕,李宏基,张梦雪,张天勇,樊少武,周金华,马 明,孙 鹏,吴文惠
(1.上海海洋大学食品学院,上海 201306;2.海军军医大学药学系,上海 200433;3.中国人民解放军69235部队,新疆奎屯 833200;4.中国人民解放军69245部队,乌鲁木齐 830000)
放线菌是临床抗生素的重要来源,约三分之二的抗生素都是由放线菌产生的[1]。放线菌产生的次级代谢产物具有抗菌[2-3]、抗肿瘤[4-5]和抗病毒[6]等多种生物活性。随着天然药物化学研究的迅速发展,大量抗生素被发现[7]。然而,抗生素的滥用导致多种病原菌的抗药性增强,严重威胁人类健康[8],因此迫切需要开发结构新颖、活性显著的新型候选药物。海洋放线菌是尚未被很好开发的微生物资源[9],海洋环境与陆地环境在光照、压力和盐度等方面存在很大差异[10],这使海洋放线菌成为新型次级代谢产物的重要来源[11]。目前研究最多的海洋放线菌是链霉菌属(Streptomyces)菌类,海洋链霉菌的天然产物中大约有67.3%表现出肿瘤细胞毒活性和抗疟、抗寄生虫等生物活性,具有巨大的开发前景和研究价值[12]。
在全球范围内,感染侵袭性真菌疾病的人数呈现递增趋势[13]。据统计,全球每年发生75万例侵袭性念珠菌病,并导致出现超过5万例死亡病例,白色念珠菌是侵袭性真菌疾病的主要病原菌[14]。目前,临床用于治疗真菌感染的药物主要有氮唑类、棘白菌素类、两性霉素类和核苷类,抗真菌药物相比于其他抗菌类药物,数量和种类都十分有限。同时,由于氮唑类抗真菌药物的长期使用,导致耐药菌越来越普遍;而两性霉素类药物具有明显肝肾毒性[15];棘白菌素类药物的耐药率也在增加[16],因此亟需发掘新的抗真菌药物。海洋放线菌中很多次级代谢产物具有抗真菌活性,例如ZHANG等[17]从海洋放线菌Streptomycessp.ZZ741中分离得到化合物streptovitacin A,对白色念珠菌有很强的抑制作用。本文对海洋放线菌Streptomycessp.MA12的次级代谢产物进行化学成分研究,并对单体化合物进行抗真菌活性测试,以期为开发新型抗真菌活性天然化合物提供科学参考。
1 材料与方法
1.1 材料与试剂
菌株:菌株MA12分离自中国南海西沙群岛海域的海绵样品,经鉴定属于链霉菌属(Streptomyces)。抗真菌活性测试菌株分别为标准敏感白色念珠菌SC5314、临床分离耐氟康唑白色念珠菌901、实验室基因编辑耐氟康唑白色念珠菌816。菌株MA12和白色念珠菌保存于海军军医大学药学系。
放线菌培养基:种子培养基(40 g葡萄糖,10 g大豆蛋白胨,21 g麦芽提取物,1 L去离子水,pH 7.0);发酵培养基(20 g黄豆粉,40 g葡萄糖,19.5 g MES水合物,1 L去离子水,pH 6.8)。抗菌测试培养基为RPMI 1640培养基(支原体肉汤培养基)和SDA培养基(沙氏葡萄糖琼脂培养基)。
色谱填料和试剂:MCI中压色谱凝胶,Sephadex LH-20葡聚糖凝胶(Pharmacia Biotech,Sweden),正相硅胶(100~200目和300~400目,烟台黄务硅胶开发实验厂),高效薄层层析硅胶板(TLC板,烟台黄务硅胶开发实验厂)。显色剂为10%硫酸香草醛溶液;HPLC所用甲醇和乙腈为色谱纯,上述试剂均购自泰坦科技股份有限公司。
1.2 MA12菌株中化合物分离纯化
1.2.1 菌株发酵培养
配置10瓶80 mL种子培养基,121℃高温蒸汽灭菌20 min,培养基冷却后接入MA12菌株,置于28℃、220 r·min-1条件下的摇床中,培养48 h。将种子液按照每瓶10 mL的方式接种到装有250 mL发酵培养基的1 L锥形瓶中。置于28℃、220 r·min-1条件下的摇床中,培养7 d,总发酵量20 L。
1.2.2 菌株化合物的提取分离
发酵液中加入等量的乙酸乙酯,手动搅拌均匀,以35 kHz频率超声提取30 min,重复5次,最终粗产物重量为26 g。用MCI中压柱分离粗产物。采用甲醇-水梯度洗脱(30∶70→100∶0,v∶v),每个梯度洗脱2.5个柱体积,得到M1~M15共15个部分。将M8组分(750 mg)经过Sephadex LH-20凝胶柱色谱分离(二氯甲烷∶甲醇为2∶1,v∶v)进行洗脱,TLC检测合并得到主产物组分M8L3(450 mg),用硅胶柱色谱分离M8L3,采用二氯甲烷-甲醇梯度洗脱(每个梯度洗脱3个柱体积,体积比分别为100∶0、50∶1、40∶1、30∶1、20∶1、15∶1、10∶1、5∶1、1∶1、0∶100),TLC检测合并得到S1~S15共15个组分,主产物M8L3S3组分(80.7 mg)经 HPLC(60% 甲醇,流速 2 mL·min-1)分离得到化合物2(13.4 mg,tR=32 min)。将M9组分(80 mg)经HPLC(68%甲醇,流速2 mL·min-1)分离得到化合物3(19 mg,tR=34 min)。M10经过Sephadex LH-20凝胶柱色谱分离(二氯甲烷∶甲醇为2∶1;v∶v)进行洗脱,TLC检测合并得到主产物组分M10L3(180.5 mg),将M10L3组分用HPLC(62%甲醇,流速2 mL·min-1)纯化,得到化合物1(7 mg,tR=30 min)。
1.3 结构鉴定方法
化合物结构鉴定主要采用波谱学方法,包括NMR(1H 谱和13C谱)和高分辨质谱(ESIHRMS),并结合微谱网站检索和文献数据比对等方法完成。在300 K温度条件下采集NMR数据,NMR化学位移通过百万分率(δ)表示,并以CD3OD信号(δH=3.31×10-6mg·L-1;δC=49.0×10-6mg·L-1)或者CDCl3信号(δH=7.26×10-6mg·L-1;δC=77.0×10-6mg·L-1)作为内标。耦合常数(J)以Hz为单位。高分辨质谱数据通过质荷比(m/z)表示,以碘化钠异丙醇溶液(2 mg·mL-1)作为参照,分辨率为5 000。
1.4 化合物活性测试
采用微量液基稀释法测定化合物最低抑菌浓度(MIC)[18]。菌液用RPMI 1640培养液稀释至浓度为(1~5)×103个·mL-1备用。将化合物置于灭菌后的1.5 mL离心管中,用DMSO溶解样品制成6.4 mg·mL-1的母液(-20℃保存)。实验前将母液置于常温融化并用RPMI 1640培养液稀释10倍(640μg·mL-1),将96孔无菌板每排的1号孔加入20μL化合物溶液和180μL菌液;2~10号孔加入100μL菌液,倍比稀释配置药物浓度为64.000、32.000、16.000、8.000、4.000、2.000、1.000、0.500、0.250、0.125μg·mL-1;11号孔加入100μL培养液作为空白对照;12号孔加入不含药物菌液作为阴性对照。上述实验在无菌超净台内进行,平行操作3次。药敏板置于35℃温箱培养。培养48 h后,用酶标仪在630 nm检测OD值。
2 结果与讨论
2.1 放线菌MA12的化合物结构鉴定
通过菌株发酵、代谢产物分离及波谱学分析,并结合文献数据比对[19-21],从菌株MA12代谢产物中鉴定出3种化合物,其化学结构式如图1所示。
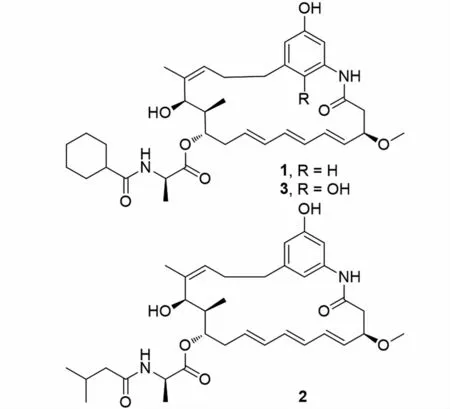
图1 化合物1~3结构式Fig.1 Structures of compounds 1-3
化合物1:白色粉末,ESI-MSm/z:623.383 3[M+H]+,分子式C36H50N2O7,不饱和度为13。其NMR 数据归属如下:1H-NMR(600 MHz,CDCl3)δH:7.59(s,1H),7.48(s,1H),6.47(s,1H),6.22(s,1H),6.10(dd,J=7.1、2.9 Hz,2H),6.03(d,J=6.8 Hz,2H),5.58(dd,J=15.3、7.2 Hz,2H),5.18(d,J=7.2 Hz,1H),4.95~4.90(m,1H),4.63(s,1H),4.40(p,J=7.0 Hz,1H),4.12(d,J=9.9 Hz,1H),3.48(s,1H),3.39(s,3H),2.76(dd,J=13.9、3.6 Hz,1H),2.59(dd,J=13.8、8.6 Hz,1H),2.51~2.47(m,3H),2.32(dd,J=14.6、8.9 Hz,1H),2.15~2.07(m,2H),1.86(d,J=10.1 Hz,3H),1.79(s,3H),1.66(d,J=11.7 Hz,2H),1.41(t,J=12.3 Hz,3H),1.35(d,J=7.1 Hz,3H),1.22(m,6H),0.92(d,J=6.8 Hz,3H)。13C-NMR(150 MHz,CDCl3)δC:176.8(C-30),173.1(C-27),168.8(C-1),157.4(C-22),144.3(C-18),138.7(C-20),138.5(C-14),134.3(C-5),133.7(C-7),133.5(C-8),130.8(C-4),129.6(C-6),129.5(C-9),125.0(C-15),112.2(C-19),111.1(C-23),106.0(C-21),78.8(C-3),75.6(C-11),68.6(C-13),56.9(C-26),48.7(C-28),45.2(C-31),43.7(C-2),39.7(C-12),36.4(C-17),33.4(C-10),29.7(C-16),29.6(C-32),29.5(C-36),25.8(C-34),25.8(C-33),25.7(C-35),20.5(C-25),17.9(C-29),10.0(C-24)。以上数据分析结合文献比对[19],确定该化合物为trienomycin A。化合物1最早是从Streptomycessp.No.83-16中分离得到,对人宫颈癌细胞有抑制作用,IC50为0.005 μg·mL-1。据文献报道,化合物1对胰腺癌细胞增殖有抑制作用[20],化合物1通过抑制STAT3转录影响癌细胞的增殖、生长、迁移和入侵,以阻滞胰腺癌细胞发展,对因STAT3异常导致的早期和晚期胰腺癌有潜在的治疗作用。
化合物2:白色粉末,ESI-MSm/z:597.351 9[M+H]+,分子式C34H48N2O7,不饱和度为12。其NMR 数据归属如下:1H-NMR(600 MHz,CDCl3)δH:7.69(s,1H),7.46(s,1H),6.47(s,1H),6.22(s,1H),6.10(dd,J=10.1、5.0 Hz,2H),6.03(dd,J=12.1、5.7 Hz,1H),5.63~5.52(m,2H),5.19(t,J=6.9 Hz,1H),4.93(dd,J=8.8、5.8 Hz,1H),4.64(d,J=4.2 Hz,1H),4.42(m,1H),4.13~4.08(m,1H),3.48(d,J=5.5 Hz,1H),3.37(s,3H),2.76(dd,J=13.7、3.6 Hz,1H),2.63~2.55(m,1H),2.51(dd,J=12.4、5.6 Hz,2H),2.47~2.40(m,2H),2.31(ddd,J=14.8、9.1、2.3 Hz,2H),2.24~2.15(m,2H),2.06(d,J=2.1 Hz,2H),1.96(s,1H),1.88(dd,J=9.1、4.3 Hz,1H),1.78(s,3H),1.37(d,J=7.1 Hz,3H),0.90~0.94(m,9H)。13C-NMR(150 MHz,CDCl3)δC:173.3(C-30),173.0(C-27),168.9(C-1),157.4(C-22),144.2(C-18),138.6(C-20),138.4(C-14),134.3(C-5),133.8(C-7),133.5(C-8),130.8(C-4),129.6(C-6),129.5(C-9),125.0(C-15),112.3(C-19),111.1(C-23),106.1(C-21),78.9(C-3),75.6(C-11),68.6(C-13),56.9(C-26),48.8(C-28),45.7(C-31),43.7(C-2),39.7(C-12),36.4(C-17),33.3(C-10),29.6(C-16),26.3(C-32),22.6(C-33),22.6(C-34),20.5(C-25),17.8(C-29),10.0(C-24)。以上数据分析结合文献比对[19],确定该化合物为trienomycin B。化合物2最早从Streptomycessp.No.83-16中分离得到,对人宫颈癌细胞有抑制作用,IC50为0.2μg·mL-1。
化合物3:白色粉末,ESI-MSm/z:639.368 2[M+H]+,分子式C36H50N2O8,不饱和度为13。其NMR 数据归属如下:1H-NMR(600 MHz,CD3OD)δH:6.57(d,J=2.9 Hz,1H),6.48(d,J=2.9 Hz,1H),6.21(dd,J=15.0、10.5 Hz,2H),6.12~6.05(m,2H),5.67(ddd,J=15.6、10.5、5.3 Hz,1H),5.50~5.43(m,1H),5.19~5.15(m,1H),4.70(m,1H),4.33~4.26(m,1H),4.12(ddd,J=10.4、8.7、4.7 Hz,1H),3.33(s,3H),2.90(dd,J=12.6、5.2 Hz,1H),2.83(dd,J=12.7、4.7 Hz,1H),2.60~2.54(m,2H),2.30(t,J=3.3 Hz,1H),2.29~2.25(m,2H),2.24~2.18(m,2H),1.92(ddd,J=13.6、6.8、3.3 Hz,2H),1.82(s,1H),1.80(d,J=5.1 Hz,2H),1.73~1.71(m,3H),1.51~1.45(m,2H),1.41(d,J=7.3 Hz,3H),1.37~1.35(m,2H),1.34(m,2H),1.32(dd,J=7.6、4.5 Hz,2H),1.30~1.26(m,2H),0.96~0.88(m,2H),0.81(d,J=6.9 Hz,3H)。13C-NMR(150 MHz,CD3OD)δC:179.2(C-30),173.9(C-27),171.5(C-1),150.7(C-22),142.0(C-19),139.6(C-14),136.6(C-5),135.7(C-7),134.7(C-8),133.2(C-18),131.1(C-4),130.7(C-9),130.6(C-6),127.3(C-20),124.9(C-15),116.3(C-23),108.4(C-21),81.7(C-3),76.5(C-11),69.6(C-13),56.7(C-26),50.2(C-28),45.9(C-31),43.6(C-2),39.6(C-12),33.8(C-10),32.6(C-17),30.8(C-32),30.7(C-36),27.6(C-16),27.0(C-35),26.9(C-34),26.8(C-33),21.0(C-25),17.2(C-29),9.7(C-24)。以上数据分析结合文献比对[21],确定该化合物为ansatrienin B。化合物3最早是从StreptomycesrishiriensisT-23中分离得到。
本文从海洋放线菌Streptomycessp.MA12分离得到3种已知安莎三烯类化合物。安莎三烯(trienomycins)属于安莎霉素类抗生素,因其结构中含有共轭三烯而得名。已经报道的该类抗生素约有200种,其结构特征是以3-氨基-5-羟基苯甲酸(AHBA)作为起始单元形成大环内酰胺[22],代表性化合物有利福霉素(rifamycin)[23]、格尔登霉 素 (geldanamycin)[24]和 美 登 木 素(maytansinoids)[25]等。安莎霉素类化合物是药物研发中的一个重要家族,具有抗结核、抗菌、抗肿瘤等生物活性。安莎霉素化合物按结构可分为萘安莎霉素类和苯安莎霉素类,萘安莎类化合物抗菌活性较好,苯安莎类化合物在抗肿瘤活性方面表现突出[26]。苯安莎类化合物的代表格尔登霉素能够与热休克蛋白Hsp90上的ATP位点结合,使Hsp90蛋白的ATPase失活而产生抗肿瘤作用[27];萘安莎化合物利福霉素对细菌RNA聚合酶具有特异性抑制作用,从而实现抗结核效果[28]。根据碳骨架的长短,苯安莎类化合物还可以分为C-15安莎霉素和C-17安莎霉素,它们在体外对肝癌细胞和小鼠白细胞等都有良好的细胞毒活性[29-30]。
2.2 化合物活性测试
选取3株白色念珠菌标准敏感白色念珠菌SC5314、临床分离耐氟康唑白色念珠菌901和实验室基因编辑耐氟康唑白色念珠菌816进行抗真菌活性测试,氟康唑(FCZ)用作阳性对照,活性测试结果如表1所示。结果显示,化合物3对标准敏感白色念珠菌SC5314、耐氟康唑白色念珠菌816、耐氟康唑白色念珠菌901均有明显抑制作用,MIC值分别为4、4、8μg·mL-1。化合物1和化合物2对3株白色念珠菌无抑制作用,MIC值均大于64μg·mL-1。本文首次报道了安莎三烯对白色念珠菌特别是氟康唑耐药菌株的抗菌活性,不仅展现了化合物3的新颖抑菌效果,还提示化合物3中的C19位羟基和环己烷侧链可能对抗真菌效果具有重要作用,后期将进一步开展构效关系和抗菌机制研究。
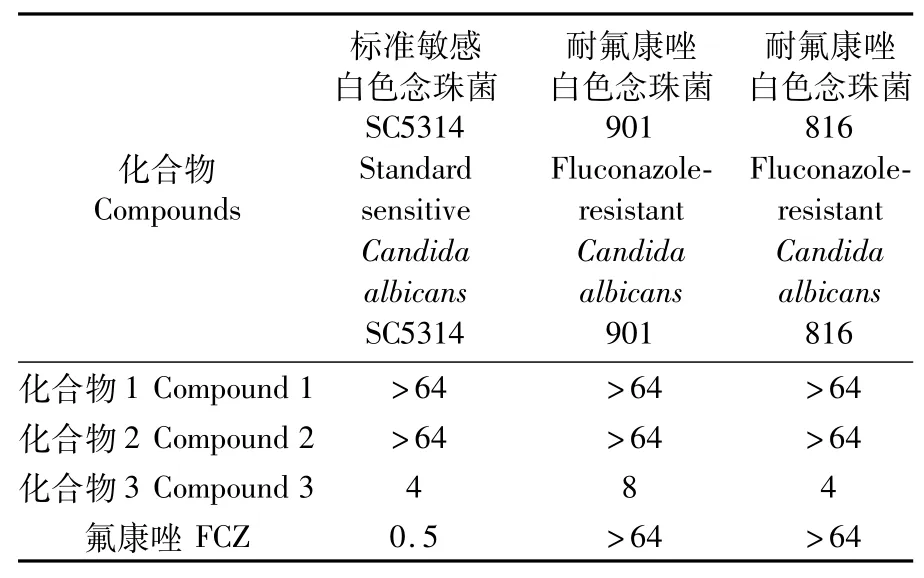
表1 MA12代谢产物的抗真菌活性Tab.1 Antifungal activities(M IC)ofm etabolites from MA12(μg·m L-1)
安莎三烯类化合物抗真菌活性测试结果为抗真菌新药研发提供了新的科学依据。本文从菌株MA12次级代谢产物中分离得到3种安莎三烯类化合物,后期可以通过改变培养基营养物质组成(碳氮源配比、微量元素、特殊代谢物添加等)、发酵条件(温度、pH、光照、时间等)以及扩大发酵规模等方式获得更多类似物。通过基因组测序可以获得菌株中与次级代谢产物生物合成相关的基因簇信息,对处于沉默状态、低表达或不表达状态的生物合成基因簇,可以利用同源重组技术替换启动子、删除抑制子,进而更高效地激活沉默基因簇,提高获得新颖活性次级代谢产物的机会。
猜你喜欢
国际呼吸杂志(2019年22期)2019-12-09
现代检验医学杂志(2016年1期)2016-11-12
兽医导刊(2016年12期)2016-05-17
西南军医(2016年1期)2016-01-23
中国卫生标准管理(2015年5期)2016-01-14
山东医药(2015年13期)2016-01-12
中国医疗美容(2015年1期)2015-07-12
现代检验医学杂志(2015年2期)2015-02-06
应用海洋学学报(2014年4期)2014-11-22
天然产物研究与开发(2014年6期)2014-04-27
