
New carbon–nitrogen–oxygen compounds as high energy density materials
2023-10-11 07:55JunyuShen沈俊宇QingzhuoDuan段青卓JunyiMiao苗俊一ShiHe何适KaihuaHe何开华WeiDai戴伟andChengLu卢成
Chinese Physics B 2023年9期
Junyu Shen(沈俊宇), Qingzhuo Duan(段青卓), Junyi Miao(苗俊一), Shi He(何适),Kaihua He(何开华), Wei Dai(戴伟), and Cheng Lu(卢成),†
1School of Mathematics and Physics,China University of Geosciences(Wuhan),Wuhan 430074,China
2Faculty of Materials Science and Chemistry,China University of Geosciences(Wuhan),Wuhan 430074,China
3School of Mathematics and Physics,Jingchu University of Technology,Jingmen 448000,China
Keywords: molecular crystals,high pressure,structure searches,first principles calculations,high energy density materials
Molecular crystals are a type of important solid-state materials in which constituent molecules are arranged in a regular, repeating pattern to form a crystal lattice.The stabilities and chemical bonding characteristics of molecular crystals are determined by the types of molecules, the crystal structures and symmetries, as well as intermolecular interactions between the molecules.For instance, under ambient conditions, molecular nitrogen gas, N2, is highly stable and inert due to the strong N≡N triple bond between N atoms,characterized by a bond length of 1.10 ˚A and a dissociation energy of 954 kJ/mol.[1]At pressures more than 110 GPa,[2–4]N–N single bonds become the preferred forms.The N≡N triple bond is much stronger than the N=N double bond (418 kJ/mol) or the N–N single bond (160 kJ/mol),[5]and the decomposition of polymeric nitrogen bonded by the single bond can release a large amount of energies.In principle, solid nitrogen and nitrogen-based compounds are promising candidates of high energy density materials (HEDMs).High pressure[2,3,6–8]is an effective method to break the extremely strong N≡N triple bonds and to yield polymeric nitrogen compounds with N–N single bonds.[9,10]In fact, many kinds of polymeric nitrogen,such as cg-N,LP-N and HLP-N are observed under high pressure.[11]However, it is still a challenge to release these high-pressure phases of nitrogen to be metastable under ambient conditions.Considering the harshness of synthetic conditions and the desirable energetic properties of polymeric nitrogen compounds,[12–17]it is still necessary to find new nitrogencontaining compounds.
One feasible avenue is to synthesize nitrogen-containing compounds through high pressure, and then release pressure to ambient pressure and stabilize them.Another possible way to achieve polymerized nitrogen compounds under ambient conditions is the combination of nitrogen with other elements through chemical pre-compression.[18–21]These processes can promote formation of polymeric nitrogen compounds and stabilize them under relatively low pressure or ambient pressure.Carbon (C), nitrogen (N) and oxygen (O) are essential elements on the Earth.They are highly abundant and readily available,and numerous compounds comprising these elements have been verified as HEDMs,[22,23]for example,carbon monoxide,nitrogen dioxide and dinitrogen pentoxide.The bond energies of multiple bonds in these compounds are 256.5 kcal/mol(C≡O triple bond),145.0 kcal/mol(N=O double bond), and 114.0 kcal/mol (N=O double bond), and the bond energies of single bonds of these elements in different compounds are about 84.0–131.0 kcal/mol(C–O single bond)and 48.0–84.0 kcal/mol (N–O single bond).In fact, several compounds such as OCNNO,CNNO2,and NCNO2are experimentally synthesized.[6,12]Although significant progress has been made by numerous researchers in recent years,a comprehensive understanding of the structural and chemical bonding characteristics relevant to the polymerization and decomposition processes of nitrogen compounds containing C and O is essential for gaining deeper insight into explosive properties of C–N–O systems.
In this work, we conduct an extensive structure search for C–N–O systems at different pressures via the CALYPSO method combined with first principles calculations.[24,25]Our findings indicate that nitrogen can readily react with carbon and oxygen at relatively low pressures(approximately 40 GPa)to form HEDMs with different compositions.We then focus on the stoichiometry of carbon-nitrogen-oxygen(CNO)compounds, and successfully identify thePbamphase of CNO under 100 GPa, along with two novel polymeric CNO compounds possessingC2/mandI¯4m2 symmetries.These three polymeric CNO compounds are potential HEDMs with energy densities of 2.30 kJ/g forPbamphase, 1.37 kJ/g forC2/mphase,and 2.70 kJ/g forI¯4m2 phase.

Table 1.Calculated elastic constants Cij (GPa)of CNO under ambient pressure and 100 GPa.

Fig.1.Calculated enthalpy differences relative to the Pbam phase for the CNO compound under high pressure from 0 to 100 GPa.
We have employed the CALYPSO method and firstprinciples calculations to predict the crystal structures of C–N–O systems at ambient pressure,50 GPa,and 100 GPa.[24,25]For each stoichiometry and pressure, we have conducted approximately 50 generations of structure searches with 30 different conformational geometries in each generation,and identified approximately 1500 distinct structures for the CNO compound.We have used the Viennaab initiosimulation package (VASP)[26]through the framework of density function theory(DFT)[27–29]combined with projector augmented-wave pseudopotentials and generalized gradient approximation with Perdew–Burke–Ernzerhof exchange correlation functions[30]to optimize the total energy of all predicted structures.To ensure the convergence of energy and forces to less than 1 meV/atom and 0.001 eV/˚A,we have adopted a densek-point of 2π×0.025 for the Brillouin zone integral and an energy cutoff of 800 eV.The mechanical stabilities of the predicted structures are evaluated via elastic constants calculated using the strain-stress method.The dynamic stabilities of each lowenergy structure is assessed using phonon dispersion curves simulated by the PHONOPY code.
The calculated enthalpies of the CNO compound under high pressure are illustrated in Fig.1.As shown in Fig.1,the CNO system undergoes the decomposition into CO2+C+N2combinations at pressures below 40 GPa.However,thePbam,C2/m,andI¯4m2 phases of CNO exhibit stable phases at pressures exceeding 50 GPa.Notably, the energy of theC2/mstructure is found to be lower in energy than that of thePbamstructure reported by Razaet al.,[1]suggesting that the energy characteristics of theC2/mphase is superior to that of thePbamphase.In the following, we will elaborate on the underlying mechanism.
At ambient pressure,the CNO system is decomposed into CO2+C+N2components.However,the high-pressure phases ofPbam,C2/m, andI¯4m2 structures are dynamically stable with weak van der Waals interactions.To exactly describe the structural and explosive properties of the CNO compound,we consider the DFT-D2 van der Waals corrections in the present calculations.In thePbamphase of CNO,the crystal structure is a layered geometry consisting of CN2O2tetrahedra with one C atom in the center.The optimized lattice constants area=8.390 ˚A,b=4.019 ˚A,c=2.431 ˚A,α=90.000°,β=90.000°,andγ=90.000°,respectively.The coordination numbers of C, N, and O atoms are 4, 3, and 2, respectively.The C–O bond lengths are found to be 1.437 ˚A and 1.419 ˚A,while the C–N bond length is 1.462 ˚A.The N–N bond length is 1.430 ˚A,which is longer than the typical bond length of N–N single bond(1.30 ˚A).For theC2/mstructure,there are six atoms per unit cell, and the calculated structural parameters area=12.642 ˚A,b=2.280 ˚A,c=2.273 ˚A,α=90.000°,β=79.625°,andγ=90.000°.The coordination numbers of C, N, and O atoms are 4, 3, and 2, respectively.The C–O bond lengths are 1.450 ˚A and 1.364 ˚A, and the C–N bond length is 1.387 ˚A, while the N–N bond length is 1.360 ˚A,which is longer than the 1.30 ˚A N–N single bond length.In theI¯4m2 phase of CNO, the predicted lattice constants area=b=2.401 ˚A,c=15.583 ˚A,α=90.000°,β=90.000°,andγ=90.000°.The C atoms are coordinated by two O atoms and two N atoms, while the N atoms are coordinated by two C atoms and one N atom,and the O atoms are coordinated by two C atoms.The bond length of C–O is found to be 1.472 ˚A,which is longer than the C–N bond length of 1.449 ˚A.The N–N bond length is 1.349 ˚A,which is greater than the 1.30 ˚A of the N–N single bond length.As such,the N atoms in theI¯4m2 phase of CNO exist in the form of single bonds.
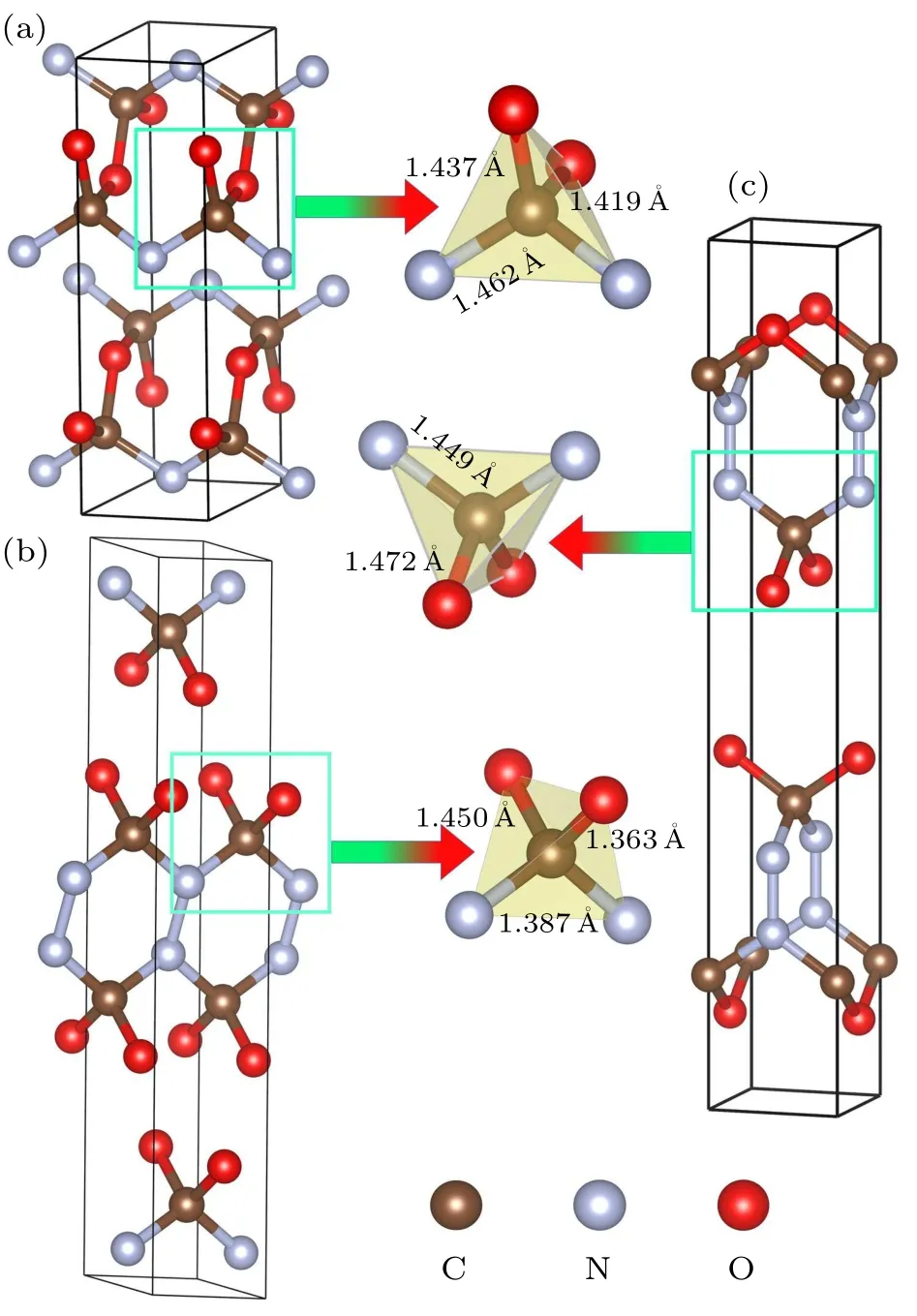
Fig.2.Crystal structures of the CNO compound at ambient pressure:(a)Pbam phase,(b)C2/m phase,and(c)I¯4m2 phase.The local tetrahedra of CNO units are illustrated for each structures.The red, brown and gray balls are oxygen,carbon and nitrogen atoms,respectively.
To confirm the dynamical stability of the CNO,we have performed phonon spectra calculations of CNO under different pressures, and the results are presented in Fig.3.The phonon dispersions of all three structures are stable under high pressure and ambient pressure, indicating dynamically stable CNO.The calculated results ofPbamphase of CNO are consistent with those reported by Razaet al.,[1]validating the reliabilities of current calculations.Additionally, the results of the electron localization function calculations of CNO reveal that bothC2/mandI¯4m2 structures exhibit typicalδbonds[31]between the C and N atoms and the C and O atoms,as well as localized electrons at the peripheries of the O and N atoms.To verify the mechanical stability of CNO, we have used theab initiostress-strain relation method to calculate the elastic constants of CNO under different pressures.

Fig.3.Phonon dispersion curves of CNO under different pressures: (a)Pbam phase at ambient pressure,(b)Pbam phase at 100 GPa,(c)C2/m phase at ambient pressure, and (d)C2/m phase at 100 GPa, (e) I¯4m2 phase at ambient pressure,(f)I¯4m2 phase at 100 GPa.

Table 2.The Bader charge analysis of CNO compounds at ambient pressure.
The calculated results are summarized in Table 1.It can be seen from Table 1 that the calculated results are perfectly satisfied with mechanical stability criteria of orthorhombic,tetragonal,and monoclinic crystals.[32]Thus,these structures are mechanically stable at ambient pressure and 100 GPa.
Based on calculations of the energy band and electronic density of states(DOS),we have found that thePbam,C2/m,andI¯4m2 phases of CNO exhibit semiconducting behavior,with corresponding band gap values of 3.18 eV,1.59 eV,and 1.32 eV, respectively.The DOSs ofPbamandC2/mphases of CNO at ambient pressure reveal that the N atoms make significant contributions to the electronic states below the Fermi level, with clear evidence of orbital hybridizations observed in all three phases.Similarly,the DOSs of theI¯4m2 phase of CNO at ambient pressure show that the N and O atoms have higher contributed values below the Fermi level.In all cases,the DOSs below the Fermi energy level are found to be dominated by C-sp,N-sp,and O-sp orbitals,indicating the presence of strong C–N,C–O,and N–N chemical bonds.Additionally,we have performed Bader charge calculations to analyze the charge transfer behaviors between different elements in CNO.The results are listed in Table 2.In theI¯4m2 structure of CNO,the Bader charge analysis[33]shows that C atoms donate about 0.63eto the N atoms and 0.80eto the O atoms.In contrast,for theC2/mstructure of CNO,the transferred electron numbers of 0.95efrom C atoms to O atoms are much larger than that to N atoms, with a value of approximately 0.30e.Based on the results of the Bader charge transition analysis, it can be concluded that the chemical bonds in theI¯4m2 structure of CNO are stronger than those in theC2/mstructure, which suggest that the energy density of theI¯4m2 structure is higher than that of theC2/mstructure.

Fig.4.Electronic properties of CNO at ambient pressure: (a) energy band and DOS of the Pbam phase, (b) energy band and DOS of the C2/m phase,and(c)energy band and DOS of the I¯4m2 phase.
In order to gain deeper understanding of the interatomic bonding interactions of the three CNO phases, we have conducted a computational analysis of the crystal orbital Hamilton population(COHP)[34,35]for these structures.The COHP analysis partitions the energy band structure, expressed in terms of orbital pair contributions,into bonding,non-bonding,and anti-bonding energy regions within a specific energy range,[36]where positive values of-pCOHP indicate the bonding energy region, and negative values indicate the antibonding region.The COHP results are presented in Fig.5.Notably,theC2/mstructure(Fig.5(b))exhibits weaker bonds compared toI¯4m2(Fig.5(c))andPbamstructures(Fig.5(a)).Additionally, the significantly larger anti-bonding states are observed belowEfin theC2/mphase, while theI¯4m2 andPbamphases lack this feature.These results are consistent with the enthalpy data for theC2/m,Pbam,andI¯4m2 phases(shown in Fig.1).The N–N bond in theC2/mstructure also displays an anti-bonding state,while no such feature is present in theI¯4m2 andPbamphases.By contrast,the strength of the C–N bonds in the three structures are not markedly different.Therefore,considering the limited number of N–N bonds,we infer that the stronger C–O bond is the primary factor contributing to the higher enthalpy of the corresponding CNO compound and resulting in the release of more energy during automatic decomposition under ambient pressure.

Fig.5.Crystal orbital Hamilton population(pCOHP)analysis of CNO:(a)Pbam phase,(b)C2/m phase,and(c)I¯4m2 phase.
To evaluate the energy density of CNO, we decompress the three high pressure phases to ambient pressure and consider the following reaction equations:
where the three metastable phases of CNO are anticipated to undergo exothermic decomposition into carbon monoxide(graphitic carbon and molecular carbon dioxide) andα-N(molecular N2)with solid carbon(graphitic carbon and molecular carbon dioxide).Using DFT calculations,we have calculated the chemical energies released during the decomposition reactions of CNO, which are 4.01 eV, 2.39 eV, and 4.69 eV for thePbam,C2/m,andI¯4m2 phases,respectively.Based on these values, we have estimated the energy densities per unit mass to be approximately 2.30 kJ/g, 1.37 kJ/g, and 2.70 kJ/g for thePbam,C2/m,andI¯4m2 phases,respectively.
In addition, we have also calculated these explosion parameters by our newly developed HEDPC method.The results are presented in Table 3.It can be seen from Table 3 that the explosive properties of CNO are analogous to modern explosives TNT,HMX,and RDX.Some parameters are better.For instance, some parameters ofI¯4m2 phase of CNO are larger than those of TNT,HMX,and RDX.This compares to values in the range of modern explosives such as TNT, HMX, and RDX,suggesting that these structures have the potential to be high energy density materials.
In summary, we have utilized a combination of the CALYPSO method and first principles calculations to comprehensively investigate the crystal structures of CNO under high pressure.We discovery two novel phases, aC2/mstructure and anI¯4m2 structure,which are found to be dynamically stable under both high and ambient pressures and exhibit significant potential as HEDMs.Specifically, theC2/mandI¯4m2 phase of CNO exhibit energy densities per unit mass of approximately 1.37 kJ/g and 2.70 kJ/g, respectively, which are comparable to those of contemporary explosives,such as TNT,RDX,and HMX.Our findings provide a detailed understanding of the structural evolution and explosive properties of CNO under high pressure,which offer important insights for designs and syntheses of new HEDMs.

Table 3.The explosive properties of ternary CxNyOz compounds and modern explosives,such as TNT,HMX,and RDX.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant Nos.12174352 and 12111530103), the Fundamental Research Funds for the Central Universities, and China University of Geosciences(Wuhan)(Grant No.G1323523065).

- Chinese Physics B的其它文章
- Dynamic responses of an energy harvesting system based on piezoelectric and electromagnetic mechanisms under colored noise
- Intervention against information diffusion in static and temporal coupling networks
- Turing pattern selection for a plant–wrack model with cross-diffusion
- Quantum correlation enhanced bound of the information exclusion principle
- Floquet dynamical quantum phase transitions in transverse XY spin chains under periodic kickings
- Generalized uncertainty principle from long-range kernel effects:The case of the Hawking black hole temperature