
UiO系列MOFs后合成金属化衍生物在非均相催化中的应用
2023-10-13 12:10魏娜,提畅,赵震,3
沈阳师范大学学报(自然科学版) 2023年3期
魏 娜, 提 畅, 赵 震,3
(1. 沈阳师范大学 化学化工学院, 沈阳 110034;2. 沈阳师范大学 能源与环境催化研究所, 沈阳 110034;3. 中国石油大学(北京) 重质油国家重点实验室, 北京 102249)
金属有机框架(metal-organic frameworks, MOFs)作为一类快速发展的新型多孔材料,在气体吸附与存储[1]、分离[2]、传感[3]、催化[4]、生物医药[5]等领域均展示出巨大的应用潜力。由无机金属原子和有机配体组装而成的MOFs材料具有模块化的结构,可通过在合成阶段有目标选择金属和配体或者对MOFs进行后合成修饰等手段对MOFs的结构和功能进行目标化设计。后合成金属化(post-synthetic metalation, PSM)就是一种MOFs的后合成修饰手段。通过PSM可将金属催化位点通过添加、 交换和封装等方式安装到MOFs材料上,既能有效提高MOFs材料的催化性能, 又能解决传统金属催化剂催化反应接触面小、 催化位点聚集失活、不便回收再利用等问题[6]。
UiO(University of Oslo)系列MOFs是由Zr6(μ3-O)4(μ3-OH)4次级结构单元(secondary building units, SBUs)和线性二羧酸配体构筑而成, 随着配体长度的变化, 分别命名为UiO-66(对苯二甲酸)、 UiO-67(4,4’-联苯二羧酸)、 UiO-68(三联苯二羧酸)、 UiO-69(四联苯二羧酸)[7]。 UiO系列MOFs的结构具有很强的可设计性, 可采用官能化的配体直接合成, 或者通过后合成手段对配体和SBUs进行处理, 都可在保证拓扑结构保持不变的同时实现结构的功能化。 同时, 该系列材料优异的结构稳定性使其能适应多种反应条件, 因而为催化应用提供了理想的体系。 本文将从配体和金属节点2个角度出发, 针对UiO系列MOFs后合成金属化的策略及在非均相催化中的应用进行系统的介绍。
1 配体上的PSM
含有能够锚定金属的开放配位点是MOFs材料通过配体实现PSM的先决条件,通过在MOFs合成步骤中选用含有第2配位点的配体(联吡啶基团、卟啉基团等)或者通过后合成配体修饰、交换等手段都可以实现MOFs中开放配位点的形成。
1.1 功能性配体引入开放性金属锚定位
1.1.1 双氮螯合位点
在过渡金属配位化学中,2,2’-联吡啶是一种被广泛应用的双齿螯合配体[8],将2,2’-联吡啶基团引入MOFs中可实现进一步的PSM。2014年,Manna等[9]以2,2-联吡啶-5,5’-二羧酸(H2bpydc)和ZrCl4为原料,通过溶剂热法合成了具有UiO-67构型的BPY-UiO。利用[Ir(COD)(OMe)]2(COD-1,5-环辛二烯)和[Pd(CH3CN)4][BF4]2通过PSM处理分别向BPY-UiO上的联吡啶螯合位点引入Ir和Pd位点(图1)。基于引入金属的特性,BPY-UiO-Ir对以双联频哪醇硼酸酯为硼化试剂的芳香族C—H键的脱氢硼化反应和苄基硅基醚的正硅化反应展示出催化活性,而BPY-UiO-Pd能高效催化取代环己酮脱氢制苯酚反应的进行。研究发现,与类似的均相联吡啶金属(Ir和Pd)配合物相比,BPY-UiO-Ir和BPY-UiO-Pd的催化活性显著提升(至少1 250倍),这归功于通过PSM引入的金属位点可以锚定在MOFs框架中固定的开放性配位点上,可实现单金属催化位点的隔离分散,有效避免活性位点聚集失活,因而展示出更高的催化活性。同时,基于UiO框架的单金属催化剂BPY-UiO-Ir和BPY-UiO-Pd也表现出高稳定性,可以循环利用。该工作为设计开发高效、稳定、可循环利用的MOFs/单金属位点催化剂提供了思路。
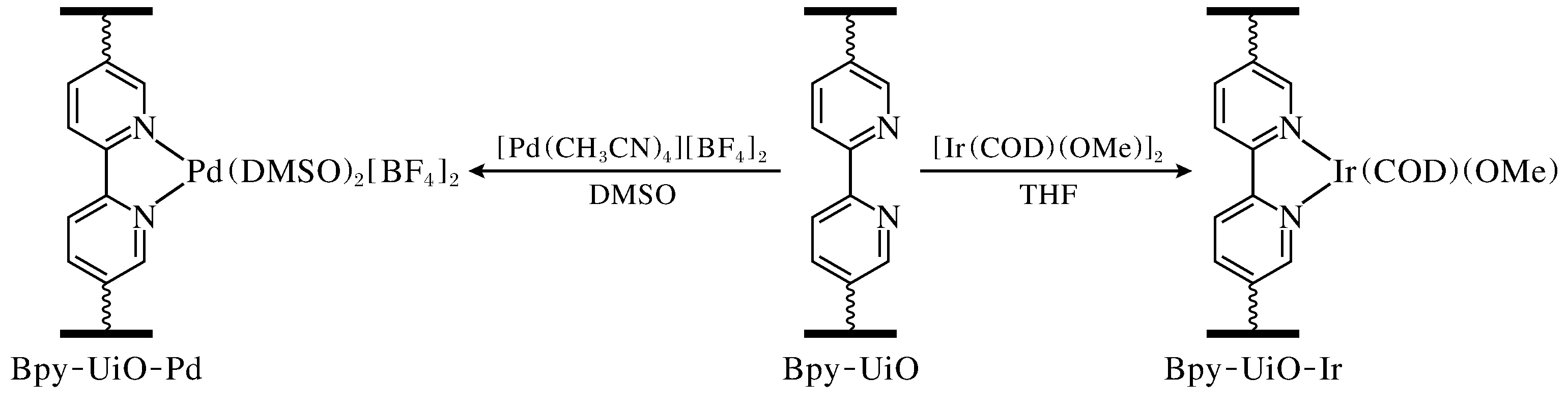
图1 BPY-UiO-M(M=Pd, Ir)的合成路线[9]Fig.1 Preparation route of BPY-UiO-M(M=Pd, Ir)[9]
2015年,Gonzalez等[10]的工作与上面类似,利用PSM制得(Ir(COD)BF4)功能化的UiO-67-bpydc。研究发现,在对苯与频哪醇硼烷和双联频哪醇硼酸酯的C—H硼化反应中,向反应体系中加入三苯基膦(triphenyl-phosphine, PPh3)或三环己基膦(tricyclohexylphosphine, PCy3)等体积较大的配位剂,可使催化剂表面的Ir中毒,导致硼化反应受到明显抑制,证实了催化主要发生在MOFs材料表面,说明Ir只与表面的联吡啶螯合而不与孔内的联吡啶螯合。
2017年,Li等[11]将25%的H2bpydc与联苯二甲酸(H2bpdc)作为混合配体与ZrCl4合成制备出UiO-67-bpy0.25,并通过PSM得到UiO-67-Co(bpy)0.25和UiO-67-Mn(bpy)0.25。这2种MOFs催化H2O2分解成水和氧气,产生的气体可以推动催化剂产生运动,可用作燃料溶液中的微型马达,而未金属化的UiO-67-bpy0.25和金属处理的不含联吡啶配位点的UiO-67在H2O2溶液中均没有运动。研究发现,该UiO-67型微型马达的运动速度可通过改变所修饰金属的种类来进行调节,其本质就是催化活性得到改变。同时,发现螯合剂亚氨基二乙酸或二乙胺四乙酸等可作为微型马达的化学刹车,通过与MOFs争夺金属离子,可达到减慢或停止反应的效果,并且制动能力与螯合能力成正比。该工作将MOFs材料的功能与自行式微/纳米机械相结合,促进了主动传输在催化、能量存储和转换、环境净化等应用中的实施。
除了联吡啶基团, 邻二氮杂菲也是一种含有开放螯合位点的官能团, 可通过配体将其引入MOFs中, 为后续金属化提供金属锚定位点。 2015年,Manna等[12]合成了2种分别含有2,2’-联吡啶基团和邻二氮杂菲基团的大尺寸线性二羧酸配体H2BPV和H2PT, 并利用这2种配体合成了UiO型MOFs, 进一步通过PSM得到3种Ir功能化的MOFs, 分别为BPV-MOF-Ir,mBPV-MOF-Ir和mPT-MOF-Ir(图2)。 MOF-Ir既具备UiO系列MOFs的高稳定性, 大尺寸线性配体又使得MOFs展示出较大的开放性孔道,从而有利于催化反应底物和产物的扩散, 同时通过实验也证实, 相较于类似的均相联吡啶基和邻二氮杂菲基Ir配合物, MOFs的框架结构锚定了Ir催化位点, 有效避免了金属位点的聚集失活, 致使MOF-Ir拥有高分散的单金属催化位点, 对一系列基于C—H活化的硅氢化、硼化偶联反应都展示出高催化活性。 该工作为开发基于氮供体配体的MOFs催化剂用于精细化学品实际合成提供了思路。

(a) BPV-MOF-Ir; (b) mBPV-MOF-Ir; (c) mPT-MOF-Ir图2 结构示意图[12]Fig.2 Schematic illustration[12]
1.1.2 卟啉基团
由于卟啉基团对多种金属有很强的配位能力,金属卟啉在催化中也有着广泛应用。因此,利用卟啉类配体构筑MOFs,也是向MOFs中引入金属锚定位的一种方式。
2018年,Liang等[13]利用含咪唑基团的对苯二甲酸(Im-H2BDC·HCl·H2O)和卟啉类四羧酸配体(H4TCPP),通过一步合成法将咪唑基团和卟啉基团引入UiO-66框架中,得到TCPP⊂Im-UiO-66。进一步通过一个连续的后合成咪唑基离子液体功能化和卟啉基金属化策略,将亲核性Br-和强Lewis酸性Zn(Ⅱ)中心引入MOF中,二者协同作用,可有效地催化环氧化物与CO2环加成转化为相应的环状碳酸酯。对于烯丙基缩水甘油醚的CO2环加成反应,TCPP⊂(Br-)Etim-UiO-66催化下产物选择性为65%,而金属化后的ZnTCPP⊂(Br-)Etim-UiO-66可将选择性提升至92%。
1.1.3 其他螯合位点
2014年, Manna等[14]通过Suzuki偶联、 醛胺缩合等多步反应合成了一种含水杨醛亚胺(sal)基团的线性三联苯二羧酸配体, 并利用该配体合成了具有UiO-68构型的sal-MOF。 sal基团的N,O原子是潜在的金属螯合位点, 因而sal-MOF经过进一步金属化处理可得到sal-M-MOF(M=Fe,Co)(图3), 它是2种烯烃加氢反应的高活性、 可回收再利用的单位点固体催化剂。 形成sal等基团的醛胺缩合, 除了可以发生在配体合成阶段, 还可以在MOFs后合成配体修饰阶段来进行, 后面将进一步讨论。

图3 sal-MOF的结构及金属化路线[14]Fig.3 Structure and metalation route of sal-MOF[14]
2018年,2种UiO-68型MOFs材料UiO-68-PCAT和UiO-68-NCAT[15]分别由含邻苯二酚和2,3二羟基萘基团的配体与其他配体混合构筑而成(图4)。配体上的邻位二羟基可螯合金属位点。其中,邻苯二酚基团具有氧化还原性,Cu(Ⅱ)金属源与其金属化作用被还原为Cu(Ⅰ),并伴随半醌自由基的形成。对于环己烯烯丙基氧化反应,含有45%Cu(Ⅰ)物种UiO-68-PCAT-Cu表现出更高的催化活性。这项工作揭示了金属—载体的氧化还原相互作用在催化剂活性中的重要性,而这种作用在传统的金属氧化物载体上很难实现。
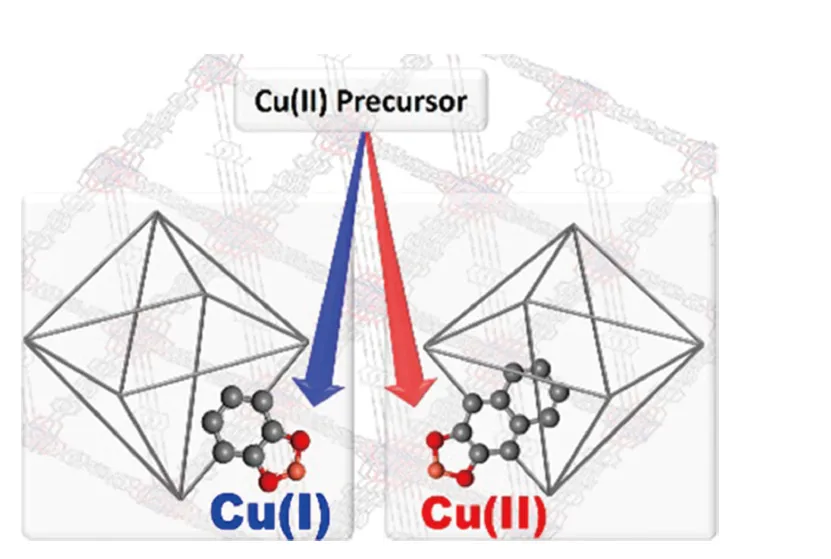
图4 金属化UiO-68-PCAT和UiO-68-NCAT结构示意图[15]Fig.4 Schematic illustration of the metallized UiO-68-PCAT and UiO-68-NCAT[15]
1.2 后合成配体修饰引入金属锚定位
后合成配体修饰是在MOFs中构筑目标官能团的重要手段[16]。通过科学选择配体,将—CHO,—NH2等官能团引入MOFs框架,再通过有机反应对其进行化学修饰,进而在MOFs中创造开放的金属锚定位。
β-diketiminate(NacNac)是一种经典双齿配体,结构中的2个N原子可与多种金属螯合[17-18],将其引入MOFs中,可实现进一步的PSM。2016年,一种由含—NH2的四联苯二羧酸配体(H2TPDC-NH2)合成的UiO-69型MOF(TPDC-NH2-UiO)[19]被报道,并利用4-苯基氨基-3-戊烯-2-酮与框架上的—NH2进行醛胺缩合反应,在MOF中构筑了NacNac螯合点(NacNac-MOF),并进一步实施了Fe,Co,Cu金属化,得到的Fe(Ⅱ)和Cu(Ⅰ)单金属位点MOFs是分子内sp3C—H氨化反应的高效催化剂,而Co(Ⅱ)单金属位点MOFs对烯烃加氢反应展示出良好的催化活性。
2021年,Antil等[20]也是通过醛胺缩合反应,将L-缬氨醇((S)-2-氨基-3-甲基-1-丁醇)嫁接到UiO-67-CHO和UiO-68-CHO上,得到手性vol-UiO MOFs。在LiHMDS(LiN[Si(Me)3]2)和FeCl2的处理下,Fe位点被亚胺N原子和去质子化的羟基氧原子螯合,得到vol-UiO-67-FeCl和vol-UiO-68-FeCl。经过LiCH2SiMe3和(OEt)2MeSiH活化处理后,结构中形成Fe-H物种(vol-UiO-FeH),在酮的不对称硅氢化反应中,反应物的羰基插入F-H并进一步与Fe配位是反应的转化限制步骤。同时,活化后的vol-UiO-Fe对脂肪族酮和芳香族酮的不对称硼氢化反应展示出优异的催化活性。
同年,Newar等[21]利用氨基酸和2-吡啶甲醛,通过串联的酰胺缩合和醛胺缩合反应,向UiO-68-NH2上修饰了一种同时含有仲胺、亚胺、吡啶基团的官能团(图5)。进一步PSM,引入的Fe位点被MOF嫁接官能团中的仲胺N、亚胺N和吡啶N三齿螯合,同时与一个THF和Cl形成5配位。得到的单位点Fe催化剂对羰基化合物的硅氢化和硼氢化反应展示出良好的活性和对映体选择性。
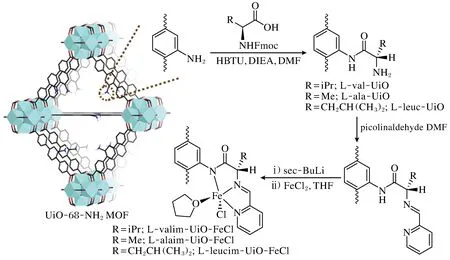
图5 UiO-68-NH2PSM路线[21]Fig.5 Post-synthetic metalation route of UiO-68-NH2[21]
1.3 配体交换引入金属锚定位
2014年,Fei等[22]通过后合成配体交换(post-synthetic exchange, PSE)将邻苯二酚基团引入UiO-66中,合成了UiO-66-CAT。邻苯二酚基团作为螯合位点可进行进一步的金属化处理,与Fe(ClO4)3和K2CrO4作用得到UiO-66-FeCAT和UiO-66-CrCAT(图6)。其中UiO-66CrCAT对一系列以TBHP或H2O2为氧化剂的醇氧化制酮反应,可以通过较低的金属负载量(0.5~1.0 mol%)催化得到高产率,而在相同条件下,UiO-66,UiO-66-CAT(PSE)及K2CrO4溶液处理过的UiO-66均展示出较差的催化效果,表明PSM引入的Cr在催化中起关键作用。
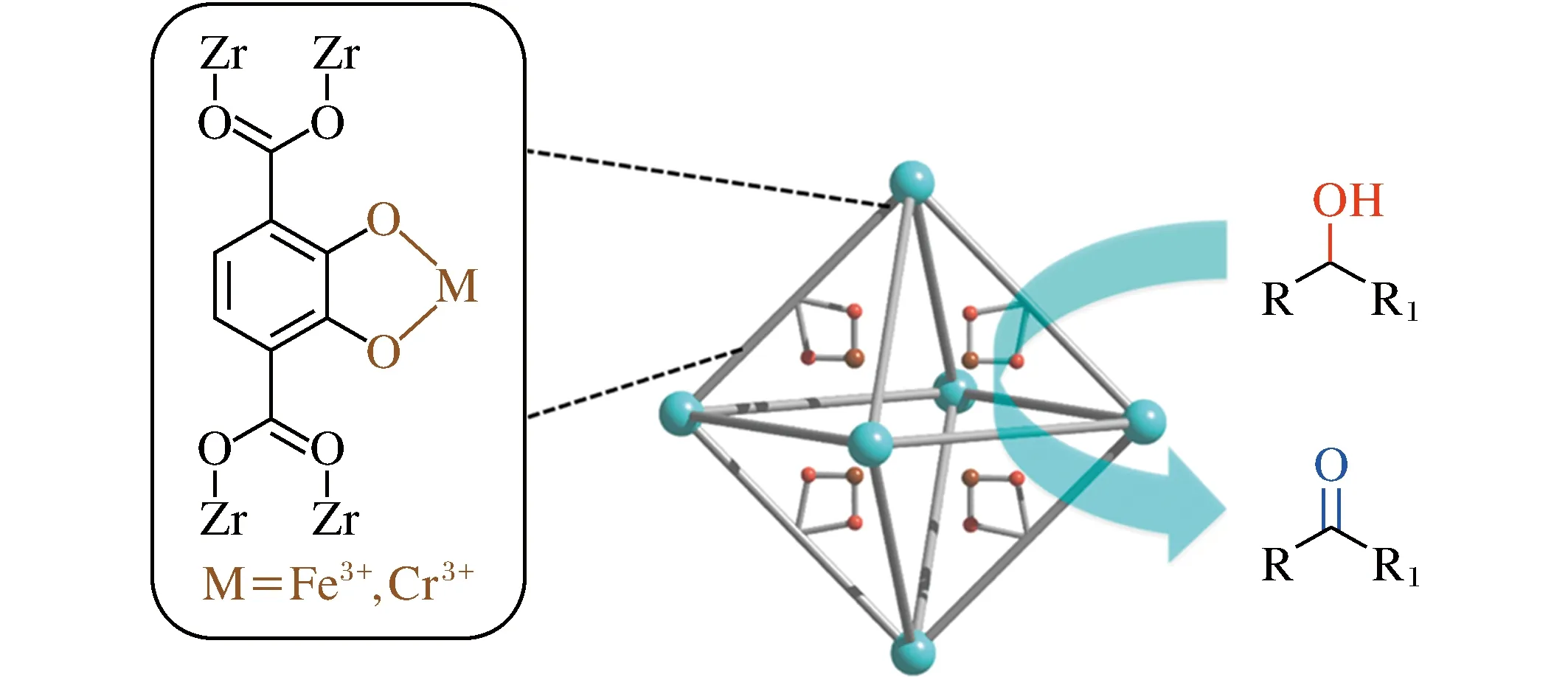
图6 UiO-66-FeCAT和UiO-66-CrCAT结构示意图[22]Fig.6 Schematic illustration of UiO-66-FeCAT and UiO-66-CrCAT[22]
2015年,该课题组同样采用PSE手段将邻苯二巯醇基团引入UiO-66骨架中,得到UiO-66-TCAT[23]。配体上2个相邻的巯基经过金属化处理将Pd位点螯合锚定在MOFs骨架上,得到UiO-66-PdTCAT。由于Pd活化C—H键的特性[24],UiO-66-PdTCAT对官能团导向的sp2C—H键功能化反应展示出催化活性。对于PhI(OAc)2作氧化剂,螯合定向氧化苯并[h]喹啉C—H键上烷氧基反应,以及N-卤代琥珀酰亚胺作为氧化剂和卤化剂的C—H键卤化反应,UiO-66-PdTCAT催化产率达95%以上,而不含Pd的UiO-66和UiO-66-TCAT催化上述反应产率均为0。
与联吡啶、卟啉基团等通过配位作用完成金属化不同的是,邻苯二酚基团和邻苯二巯醇基团是通过共价键锚定金属原子,这样的结合方式使得到的MOFs基金属催化剂更加稳定,有效克服了金属活性位点浸出的问题。
2 金属节点上的PSM
MOFs中除了配体可以作为PSM的位点,金属/金属簇SBUs也由于其种类、结构的多样性以及金属离子功能化的便利性,为MOFs材料的PSM提供了平台。
2016年,Manna等[25]合成了具有UiO-69结构的TPHN-MOF, 在室温下利用二甲基镁(Me2Mg)对其进行金属化,将Mg位点修饰在Zr—O簇的μ3-OH上,得到TPHN-MOF-Mg。该材料对醛/酮的硼氢化反应、亚胺的硼氢化反应和氨基烯烃的氢胺化反应均表现出优异的催化活性。这是第一例用于有机转化的MOFs基单位点主族金属催化剂。同年,该课题组分别用FeBr2和CoCl2溶液对UiO-68进行了金属化,得到UiO-CoCl和UiO-FeBr(图7)[26]。引入的Co(Ⅱ)和Fe(Ⅱ)中心同样是修饰在了Zr—O簇SBUs上,分别与1个μ3-OH氧原子、2个羧基氧原子和1个卤素原子形成四配位。UiO-CoCl经过NaEt3BH处理得到含有μ4-O—Co(H)组分的UiO-Co,其对苄基C—H键的化学选择性硼化和硅化,以及烯烃和酮的加氢和硼化等有机转化反应展示出良好的催化性能,而UiO-FeBr可用于催化sp3C—H键胺化。相较于配体官能团固定金属位点,将金属位点固定在MOFs金属节点上,可以避免配体对其催化活性的影响,同时也免去了设计含特定官能团配体并将其引入MOFs结构中的复杂过程,为制备MOFs基单位点金属催化剂提供了一种简单、廉价且有效的策略,为MOFs催化剂走向实际应用提供了可能性。

图7 UiO-68后合成Co功能化路线[26]Fig.7 Post-synthetic Co-functionalization route of UiO-68[26]
3 结论与展望
PSM作为一种制备功能化MOFs的有效策略,通过合理选择用于PSM的金属源,不仅可以根据金属中心的特性,而且还可以根据配位的辅助配体来制备广泛的催化活性物种,从而设计具有独特反应活性和选择性的催化材料。目前,PSM在MOFs及其衍生物的催化应用探索方面得到广泛应用,但仍面临一些挑战,例如,探索提高金属化效率的一般策略,通过建立金属化物种精确结构表征方案来深入探究活性中心的信息,进而加深对其催化应用的理解等。随着PSM技术的发展,MOFs材料在催化乃至其他方面的应用将更加广泛。
猜你喜欢
食品工业科技(2023年4期)2023-02-14
中国食品学报(2019年10期)2019-11-12
有色金属材料与工程(2017年2期)2017-05-31
材料科学与工程学报(2016年5期)2016-02-27
化学工业与工程(2016年3期)2016-02-04
中国洗涤用品工业(2015年2期)2015-02-28
郑州大学学报(理学版)(2014年3期)2014-03-01
无机化学学报(2014年12期)2014-02-28
无机化学学报(2014年9期)2014-02-28
无机化学学报(2014年3期)2014-02-28
