
灌木柳耐盐SNP位点的快速鉴定与标记开发
2023-12-04 00:46教忠意田雪瑶郑纪伟王保松何开跃何旭东
南京林业大学学报(自然科学版) 2023年5期
教忠意,田雪瑶,,郑纪伟,王保松,何开跃,何旭东*
(1.江苏省林业科学研究院,江苏省农业种质资源保护与利用平台柳树资源圃,江苏 南京 211153; 2. 南京林业大学生物与环境学院,江苏 南京 210037)
柳树属杨柳科(Salicaceae),是柳属(Salix)和钻天柳属(Chosenia)树种的统称。全世界有柳树500余种,主要分布于北半球寒带和温带地区。中国有柳树257种122个变种33个变型,其中约2/3为灌木柳类型[1]。灌木柳由于其形态各异,皮色和花芽变异丰富,常用于景观绿植与切花;其枝条通直、韧性好、青条产出率高,还被用于柳编出口海外[2];灌木柳生长速度快、适应性强、更新和萌蘖容易、光能利用率高,且对环境有很强的修复与净化作用,在欧洲和北美被广泛用于短周期生物能源林建设中[3-4]。此外,与杨树及其他乔木柳类型相比,灌木柳基因组大小适中、无性繁殖容易、世代周期短,1年即可开花,在林木遗传学和基因组学研究中极有潜力并成为新的模式树种[5]。
传统的常规育种是利用现有的自然变异或杂交手段创造变异,通过对目标性状进行表型选择达到遗传改良的目的。对于林木而言,此过程周期漫长、准确率较低且受环境影响较大。而标记辅助选择(marker-assisted selection, MAS)是通过检测与目的基因紧密连锁的分子标记达到选择目标性状的目的,极大地缩短了育种年限、提高了育种效率、加快了育种进程[6]。国内柳树分子生物学研究起步较晚,国外柳树QTL定位的研究主要集中于生长与生物量[7-9]、物候[10-11]、非生物胁迫[12-13]以及抗叶锈病[9,14-15]等性状,而与目标基因紧密连锁的功能标记开发研究仅见生长与物候相关基因[16]、平茬响应基因[17]以及性别决定基因[18-20]等报道。这些功能标记的开发主要通过连锁图谱或关联分析,通常需要大量的群体材料。近年来,混合群体分离法(bulked segregant analysis, BSA)作为一种简单、快速、有效的分子标记与目标性状紧密连锁的鉴定方法,已经在作物上得到大量的应用[21]。该方法主要选择双亲群体分离后代中具有极端表型的个体进行混样,通过比较不同极端混样池之间的多态性并结合表型进行目标基因定位。
随着高通量测序平台的不断更新,基于全基因组限制性酶切位点的简化基因组测序技术,如限制性酶切位点相关DNA测序(restriction site-associated DNA sequencing,RAD-Seq)、基因分型测序(genotyping-by-sequencing,GBS)、特定位点个段扩增测序(specific-locus amplified fragment sequencing,SLAF-Seq)等可为分子生物学的研究提供大量的多态性分子标记,再结合BSA(如SLAF-BSA)分析能够显著提升候选基因鉴定的效率,并广泛应用于各种作物[22-27]和林木[28-29]的研究中。但在柳树研究中,鲜见应用此种方法的报道。鉴于此,本研究基于SLAF-BSA技术,利用耐盐和敏盐2个灌木柳亲本及其杂交子代构建极端性状混池,通过关联分析鉴定与耐盐性状紧密连锁的SLAF标签,同时对标签序列进行功能注释,开发特异分子标记并进行验证,旨在为柳树功能基因的定位与克隆,以及分子标记辅助育种工作提供理论依据和技术支持。
1 材料与方法
1.1 试验材料
利用前期筛选的1种敏盐灌木柳欧洲红皮柳(Salixpurpurea)作母本,1个耐盐灌木柳苏柳品种‘2345’(S.×jiangsuensis‘J2345’)作父本进行杂交,共获得杂交子代3 000株。待子代小苗株高至30 cm左右时,将其从穴盘内移栽至大田进行定植。
1.2 混池构建与DNA提取
从杂交群体中选取生长旺盛、株高相近、地径较粗(1 cm以上)的单株1 505株并进行编号,采集每单株叶片放入自封袋中,置于冰箱冷藏保存。将茎干截成长度12 cm的插穗放入32孔穴盘中,每孔放同一单株的3根插穗,共设置3次重复。将穴盘置于塑料周转箱中,加入1/2 Hoagland营养液进行水培,每3天更换1次营养液,培养20 d 至根毛及叶片大量萌发。花房室温保持20~25 ℃,湿度保持70%~75%。分别加入质量分数0.1%、0.3%、0.5%和0.7%的NaCl溶液进行盐胁迫处理,以叶片颜色变化(由淡黄至枯黄)及脱落程度(从不脱落至完全脱落)为标准,筛选出极端耐盐单株50株、极端敏盐单株50株。挑选出对应编号单株的叶片,利用改良CTAB法提取DNA[30]。分别将50株耐盐单株和50株敏盐单株DNA等量混合,构建耐盐混池和敏盐混池。同时提取两个亲本DNA,利用Nanodrop将亲本和两个混池DNA质量浓度统一调整至100 ng/L,-20 ℃保存备用。
1.3 文库构建与SLAF测序
以毛果杨(Populustrichocarpa)(http://www.ncbi.nlm.nih.gov/assembly/GCF_000002775.3)基因组序列为参考基因组进行酶切预测,最终确定限制性内切酶组合为Hae Ⅲ + Hpy166 Ⅱ。分别对两个亲本和两个混池DNA进行酶切,在酶切片段3′端加A并连接Dual-index测序接头,经PCR扩增、纯化、混样后挑选长度在314~364 bp的片段作为SLAF标签进行高通量测序。测序平台为Illumina HiSeqTM2500,选用水稻‘日本晴’(Oryzasativa‘Japonica’)基因组为对照进行质控。
1.4 标记开发与关联分析
利用软件Bwa[31]将亲本与子代混池测序产生的阅读子(reads)与参考基因组进行比对,筛选亲本中具有多态性的SLAF标签和有reads覆盖区域的SNP。SNP标记的开发利用GATK[32]软件。采用SNP-index法[33]通过计算混池间显著差异基因型频率的Δ(SNP-index)指数进行标记与目标性状的关联分析,计算方法如下:
ISNP-index(aa)=Maa/(Paa+Maa);
ISNP-index(ab)=Mab/(Pab+Mab);
Δ(SNP-index)=ISNP-index(aa)-ISNP-index(ab)。
式中:Maa表示aa群体来源于母本的深度;Paa表示aa群体来源于父本的深度;Mab表示ab群体来源于母本的深度;Pab表示ab群体来源于父本的深度。将各个标记的Δ(SNP-index)从大到小排列,取99百分位数作为阈值,大于该阈值的标记则为与性状显著关联的标记。
1.5 功能注释与标记验证
将关联分析筛选出的候选SLAF标签序列与NCBI中的非冗余核酸序列(Nt)、蛋白直系同源簇(clusters of orthologous groups,COG)、直系同源蛋白分组(non-supervised orthologous,NOG)、经注释的蛋白质序列(Swiss-Prot)和KEGG(Kyoto encyclopedia of genes and genomes)等数据库进行比对,注释其功能。利用软件Primer 3[34]对SNP位点进行引物设计并交北京百万克生物科技公司合成。随机选取12个子代(6个耐盐个体,6个敏盐个体),连同2个亲本DNA为模板用于PCR反应,并进行重测序验证。PCR为25 μL反应体系,包含DNA模板1 μL,前后项引物各0.6 μL,2 ×TaqPCR Master Mix 12.5 μL,ddH2O 10.3 μL。PCR反应程序为:94 ℃预变性5 min;94 ℃变性30 s,60 ℃退火30 s,72 ℃延伸1 min,30个循环;72 ℃延伸10 min。PCR产物交北京百万克生物科技公司进行Sanger测序,利用DNAMAN 5.2.2 (Lynnon Biosoft, Quebec, Canada)进行测序序列对比。
2 结果与分析
2.1 样本SLAF标签开发及分析
1)SLAF标签统计。对母本个体、父本个体、耐盐混池以及敏盐混池共4个样本进行SLAF高通量测序,统计结果如表1所示。

表1 4个样本SLAF标签统计
测序得到的reads数变化范围为6 961 957~15 854 156个,最高为敏盐混池样本,最低为父本个体样本,平均为11 076 280个。4个样本碱基质量Q30值变化范围为93.47%~94.96%,平均为94.36%,表明测序质量较好。GC含量最高为父本个体(39.82%),最低为母本个体(38.82%),平均为39.39%。酶切共开发175 468个SLAF标签,其中敏盐混池SLAF标签数最多,为159 448个,母本个体SLAF标签数最少,为98 595个。每个样本的测序深度从35.61×至57.93×不等,平均为48.57×。
2)标签多态性分析。对4个样本中开发得到的175 468个SLAF标签根据等位基因数和基因序列之间的差异进行多态性分析,共得到3种类型的标签:多态性SLAF标签、非多态性SLAF标签以及位于重复序列区的SLAF标签。各类型SLAF标签结果表明多态性标签数量为25 675个,占标签总数量的14.63%;非多态性标签为147 339个,占标签总数量的83.97%;剩余的2 454个标签位于重复序列区,占1.4%。
3)关联分析。对多态性的25 675个SLAF标签依据亲本基因型进行筛选,去除两个亲本测序深度低于5×的标记,同时对每个标记等位基因的亲本来源依据测序信息进行确定,最终得到1 774个来源于父母本基因型的多态性SLAF标签用于后续关联分析。利用SNP-index法计算混池间显著差异基因型频率的Δ(SNP-index)指数,以99百分位为阈值,筛选出与耐盐性状显著关联的SLAF标记18个(表2)。18个标签序列共包含25个SNP位点,其中marker 116156和marker 88668包含3个SNP位点,marker 118929、marker 108616和marker 68843分别包含2个SNP位点,其余序列包含1个位点。
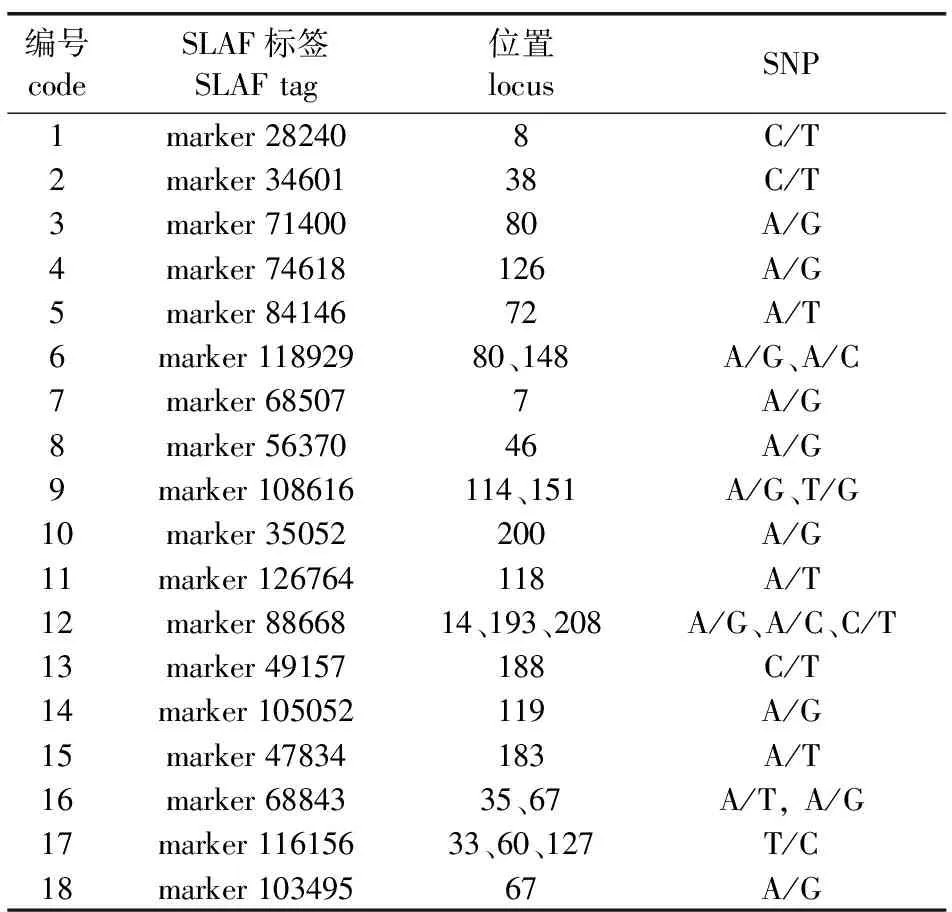
表2 耐盐相关SLAF标签统计
2.2 SLAF标签基因功能注释
对18个SLAF标签序列进行多个数据库的功能注释,其中6个标签序列产生比对结果。如表3所示,6个标签序列在Nt数据库中均有注释,且功能均与毛果杨或胡杨相关;4个标签序列marker 84146、marker 105052、marker 103495和marker 56370在NOG数据库中有注释,功能分别与翻译后修饰、转录、脂质运输和新陈代谢以及信号转导机制相关,3个标签序列marker 105052、marker 103495和marker 56370在KEGG数据库中有注释。所有的候选标记中,仅有marker 103495标记在Swiss-Prot数据库中有注释,功能与线粒体酰基载体蛋白相关。在COG数据库中没有产生比对结果。

表3 SLAF标签基因注释
2.3 SNP位点验证
利用18个SLAF标记序列针对25个候选SNP位点进行引物设计,共设计出10对引物(表4)。部分序列由于SNP位点过于靠近两端,或者没有合适的结合位点,无法进行引物设计。随机挑选6个耐盐子代和6个敏盐子代,连同父母本一并进行PCR扩增。利用琼脂糖凝胶电泳检验PCR产物并进行Sanger重测序,将测序结果进行多重序列比对。结果表明10对引物均获得PCR产物,且SNP位点的重测序结果均与原SLAF测序结果一致。标记marker 74618重测序序列比对结果见图1。如图1所示,在父本、耐盐子代个体1114、1205、194、690、44和622中,SNP位点为碱基G;在母本、敏盐子代1130、42、1884、311、2和1784中,SNP位点为碱基A。该位点在耐盐个体和敏盐个体间发生了碱基A/G的替换,与SLAF测序结果相符(表2)。由此可见,这10个标记中的SNP位点与耐盐性状紧密连锁,可以为灌木柳耐盐性的快速鉴定提供一定的参考。

图1 Marker 74618重测序序列比对结果Fig. 1 Analysis of re-sequencing sequences alignment for marker 74618

表4 SNP引物信息
3 讨 论
本研究利用SLAF-BSA技术通过关联分析快速鉴定了与耐盐性状紧密连锁的18个SLAF标签,并开发了10对SNP标记引物,相比传统依赖于连锁图谱的QTL定位研究方法,大大降低了成本、节约了时间、提升了效率。尽管混合群体分离法在遗传学和基因组学中得到了大量的应用,具有明显的优势,但也存在一定的局限性。有研究表明BSA分析不适合用于数量性状的鉴定,如株高、产量、抗病虫害等[29]。但随着研究的进一步深入,BSA越来越多的应用于一些由主效基因控制的简单数量性状的鉴别[23]。对于不同效应基因控制的数量性状,也可以通过精确的表型鉴定和环境控制试验来提升BSA分析的效率[35]。而对于由多个微效基因控制或受环境影响较大的复杂性状的鉴定,BSA分析很难实现[36]。由此可见,表型的准确鉴定是BSA分析的前提。对于容易受环境影响的表型,应通过严格的试验方法进行控制,将一些非遗传性因素如气候、土壤、区位、生物或非生物胁迫等的影响降到最低[35]。
此外,BSA分析的有效性很大程度上还与取样策略相关,包括群体的大小和混池的大小。对于质量性状或者由主效基因控制的数量性状,群体和混池的大小取决于群体类型、标记间的距离、目标区域的重组率以及目标性状的遗传结构等[37];对于微效多基因控制的复杂性状,还应考虑到基因的数量、基因的效应、基因间互作以及基因间相对位置等[38]。通常认为:对于较大效应的QTL,群体大小可控制在200~500个体,每个混池理想的个体数应占群体总数的20%~30%,至少保持在20个以上;对于中等效应的QTL,群体大小可控制在500~1 000个体,每个混池个体数保持在50个以上;对于微效QTL,群体大小可控制在3 000~5 000个体,每个混池个体数至少在100个以上[21]。本研究中,经过初步筛选,得到了包含1 505个体的群体,并分别构建了包含50个体的极端混池,基本可以满足检测到中等效应QTL位点的需要。
本研究利用18条耐盐相关的SLAF标签序列进行功能注释,其中6条序列产生比对结果。其中,marker 16156与毛果杨己糖激酶家族蛋白功能相关,marker 74618与胡杨酰基转移酶样蛋白功能相关。己糖激酶在大多数植物中均存在,是葡萄糖分解过程中的第一个关键酶,在果糖磷酸化和葡萄糖信号转导中起重要作用。有研究表明,己糖激酶基因具有防御功能,在受到高盐、低温等胁迫时,表达量显著上调[39-40]。酰基转移酶基因主要在植物种子中调节油脂的合成。在拟南芥(Arabidopsisthaliana)中,过表达的磷脂二酰甘油酰基转移酶基因可以诱导三酰基甘油的积累,从而在一定程度上缓解逆境胁迫对细胞的伤害[41]。在紫苏(Perillafratescens)中,在高盐、低温和干旱胁迫下,磷脂二酰甘油酰基转移酶基因表达量显著上调[42]。此外,植物中甘油-3-磷酸酰基转移酶通过调节软木脂合成,以维持种皮和根表皮的正常结构,进而提高植物的耐盐性[43]。由此可见,己糖激酶和酰基转移酶基因均与胁迫应答相关,并通过一定的表达机制来抵御逆境的伤害。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
世界科学技术-中医药现代化(2020年2期)2020-07-25
车迷(2018年11期)2018-08-30
海峡姐妹(2018年3期)2018-05-09
临床医药文献杂志(电子版)(2017年11期)2017-05-17
公民与法治(2016年10期)2016-05-17
西南农业学报(2016年6期)2016-04-16
中学生物学(2016年8期)2016-01-18
法医学杂志(2015年4期)2016-01-06
计算机工程(2015年8期)2015-07-03
