
低密度脂蛋白受体相关蛋白1参与抑郁症发生的可能机制
2024-01-12 12:59万振宇肖玲王高华
神经损伤与功能重建 2023年12期
万振宇,肖玲,王高华
根据世界卫生组织统计,抑郁症占全球非致命性疾病负担的10%,在2015 年已经成为第3 大致残原因,超过一半的自杀与抑郁症有关[1-3]。关于其发病机制有很多假说,包括但不限于单胺假说、下丘脑-垂体-肾上腺轴异常、神经可塑性紊乱以及炎症等[4,5]。针对已知的抑郁症的发病机制,目前的治疗手段包括药物治疗、物理治疗和心理治疗,但并不能对大部分抑郁症患者产生持久的益处,这表明抑郁症可能仍有其它的发病机制。
有研究发现在慢性不可预见性动物应激模型中,小鼠的低密度脂蛋白受体相关蛋白1(low density lipoprotein-related protein 1,LRP1)蛋白和基因表达水平增加并且出现了明显的抑郁样行为[6],这表明LRP1 可能通过某些通路参与了抑郁症的发生发展。本文将对LPR1 的常见功能及其可能导致抑郁症的通路进行综述。
1 LPR1可能导致抑郁症的通路
1.1 LRP1促进β-淀粉样蛋白(amyloid,Aβ)的积累
Aβ是一种易聚集且有毒性的多肽,其聚集形成的斑块可以导致炎症,突触功能障碍,严重时甚至导致细胞死亡。研究表明Aβ可以通过模式识别受体(pattern recognition receptors,PRRs)激活先天免疫系统诱导炎症反应。也可以与Toll 样受体4(Toll-like receptor 4,TLR4)结合激活小胶质细胞和星形胶质细胞并诱导促炎因子的释放[7]。还可以通过诱导敏感神经元过度兴奋并驱动过度激活的恶性循环导致细胞损伤。此外,随着Aβ的积累,受体细胞的微管蛋白会逐渐出现珠化和内体渗透,最终损害突触的重塑功能,影响神经的可塑性[8]。
LRP1 是Aβ的主要内吞受体,在小胶质细胞和星型胶质细胞中大量表达。研究表明LRP1 通过摄取机制介导了Aβ的清除,其表达降低与Aβ水平升高及淀粉样蛋白沉积相关,但也有实验表明神经元细胞中的Aβ的积累依赖于LRP1 的内吞作用[9,10]。LRP1表达过量时会加速Aβ内吞,当其内化的Aβ超出了溶酶体的承受范围后则会开始大量积蓄导致斑块的出现,进而导致神经元突触传递的抑制,最终致其死亡。同时LPR1表达的增加也会导致Aβ在细胞表面过度积累进而诱导细胞变性,最终导致神经可塑性功能的紊乱。当LPR1内吞功能被抑制时,会降低神经元对Aβ的摄取从而避免了Aβ的累积和斑块的形成,可以有效防止不良结局的发生[9]。
1.2 LRP1脱落为可溶性LRP1
LPR1为单链600kDa I型跨膜受体,成熟的受体由515 kDa 和85 kDa 亚基组成,通过非共价相互作用偶联。研究发现小胶质细胞上的LRP1在脂多糖、Aβ42或去整合素和金属蛋白酶10/17(ADAM10/ADAM17)等作用下会形成脱落的LRP1(soluble low density lipoprotein receptor related protein 1,sLRP1)。在小鼠脊髓中注射sLRP1 可以诱导神经炎症,在用金属蛋白酶抑制LPR1 的脱落后,白细胞介素(interleukin,IL)-6 和IL-1β的表达明显减弱[11]。sLPR1诱导炎症的途径尚未完全明确,可能与LPR1的配体,受体相关蛋白(receptor-associated protein,RAP)有关[12]。当LPR1在体内异常脱落或过度表达(意味着可用于脱落的底物数量的增加),产生的sLPR1会使神经系统处于炎症状态并损伤神经元和神经细胞,导致神经功能的异常和可塑性的紊乱。sLRP1 具有强烈的促炎作用,可以放大并维持神经炎症。研究者还发现,在星形胶质细胞和小胶质细胞的培养物中添加sLRP1 可显著抑制肿瘤坏死因子(tumor necrosis factor,TNF)-α诱导的P83 丝裂原活化蛋白激酶(mitogen-activated protein kinases,MAPKs)和细胞外信号调节激酶(extracellular signal-regulated kinases,ERK)的激活[13]。
1.3 LRP1加剧载脂蛋白E(apolipoprotein E,APOE)4的损伤作用
APOE 是大脑中主要的载脂蛋白和胆固醇受体,APOE 有3 个亚型,其中APOE 基因的ε4 等位基因是其3 个多态性等位基因(ε2、ε3 和ε4)中最强的遗传风险因子,它与多个疾病和神经退行性变的发生密切相关[14],而LRP1是大脑中主要的APOE4的受体。有研究发现APOE4可以加剧小鼠海马中Aβ40和Aβ42的不溶性量和Aβ沉积,并诱导小鼠神经变性,但这种作用会被LPR1的缺失所逆转,这表明APOE4是通过依赖LRP1的机制加剧了Aβ的损伤[15]。
此外APOE4在神经元中的过度表达还会促进小鼠tau蛋白的磷酸化损伤正常tau蛋白功能。tau蛋白有助于调节微管的稳定性,进而维持正常的突触发生和重塑以及神经发生。但APOE和tau蛋白通常被血浆或细胞器膜分开,它们如何进行物理接触仍未完全阐明。LRP1 是tau 蛋白在大脑中扩散的关键调节因子[16],同时LRP1也是APOE4的主要转运受体。因此APOE4与tau蛋白的接触很可能是以LRP1作为中介,从而产生后续反应并导致tau蛋白的磷酸化,进而导致神经可塑性的损伤。
1.4 LPR1参与金属蛋白酶组织抑制剂(tissue inhibitor of matrix metalloproteinases,TIMPs)的清除
TIMPs 是一类蛋白质家族,通过与配体的非共价结合来抑制分解素和金属蛋白酶(A disintegrin and metalloproteinases,ADAMs)的活性,其中的TIMP-3 是ADAMs 的主要生理抑制剂[17]。有研究表明ADAM10参与了丙烯醛导致的神经炎症和损伤并在丙烯醛诱导NLRP3炎症小体这一过程中起关键作用[18]。因此,TIMP-3作为ADAM的抑制剂,在预防这些不良结局的发生上有至关重要的作用,而LRP1在TIMP-3的内化和降解中起主要作用[19]。当LPR1 过度表达时,TIMP-3 会加速降解,对ADAM10的抑制作用也随之减弱,这有助于ADAM10介导各种炎症和损伤的发生。
2 LPR1与抑郁症的关系和可能的机制
目前,LPR1与抑郁症有直接关联的证据比较有限,但部分研究仍表明LPR1可能参与了抑郁症的发生发展。例如研究者观察到在慢性不可预见性动物应激模型中,小鼠LPR1 蛋白和基因表达水平增加并出现了明显的抑郁样行为[6]。在重度抑郁症患者的脑区中发现了更高的淀粉样蛋白沉积[20],而LPR1作为主要的Aβ内吞受体很可能参与了这一结局的发生。这些研究表明LPR1 可能参与了抑郁症的发生发展,但具体机制并未阐明。我们根据LPR1的功能特征和抑郁症的发病机制,对LPR1可能导致抑郁症的通路进行总结,见图1。
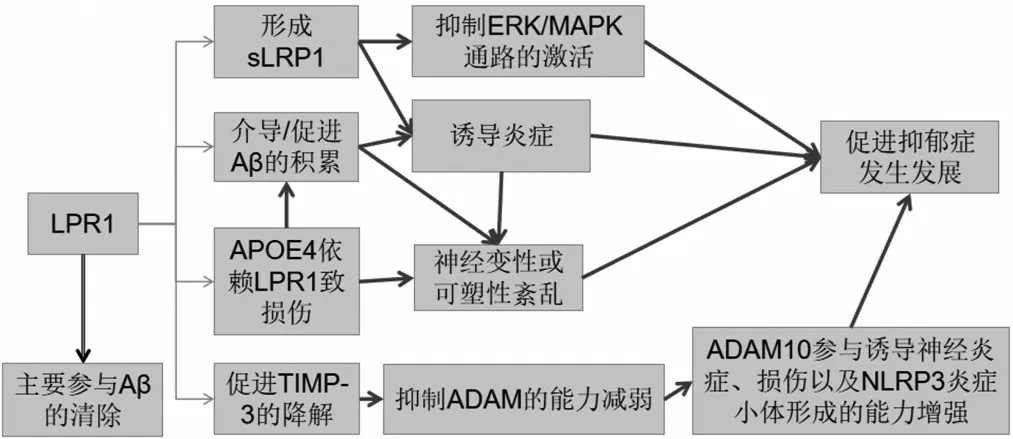
图1 LPR1可能导致抑郁症的通路
抑郁症的发生与炎症密切相关。抑郁症患者血液中的一些炎症因子与健康对照组相比明显增加,一些人类研究也表明在注射促炎物质后会出现明显的焦虑和抑郁。还有研究表明一些炎症因子,例如TNF-α和IL-6 的升高与抑郁症的非典型症状或重度抑郁症相关[21,22]。而LRP1 可以通过多种通路促进炎症的发生,例如促进Aβ的积累和斑块形成进而引起炎症反应,也可以脱落为sLRP1诱导炎症反应的发生。
神经元的损伤和神经可塑性的紊乱也是抑郁症的重要病理机制。研究表明,促进神经元发生可以有效缓解抑郁症状[23],抗抑郁药(氟西汀)可以通过逆转神经蛋白和改善神经可塑性来减少类似抑郁的行为。LPR1不仅可以通过诱导炎症损伤神经功能,还可以通过加速Aβ内吞并使Aβ在细胞表面过度积累,最终导致神经元的损伤和神经可塑性的紊乱,并且还加剧了APOE4所导致的神经损伤。
此外,当LPR1过度表达时,可以通过促进TIMP-3的降解,进而减弱对ADAM10的抑制作用,这有助于ADAM10介导的不良结局的发生。ADAM10 不仅参与了丙烯醛诱导的神经炎症和损伤,还在诱导NLRP3 炎症小体这一过程中起关键作用,许多研究已经证明NLPR3 的激活与抑郁症的发病密切相关[24,25]。因此,LPR1可能通过降低对ADAM10的抑制作用,导致不良结局并促进抑郁症的发生。
当LPR1 脱落为sLPR1 后可能也会促进抑郁症的发生。sLPR1 可以抑制ERK/MAPK 通路的激活,而ERK/MAPK 途径与情绪调节有关,特别是ERK通路在抑郁症的发病机制中有重要作用。在人类和动物抑郁症模型中,前额叶皮质和海马体的ERK信号传导都显著下调,抗抑郁药也可以通过激活ERK改善抑郁行为[26]。LPR1 过度表达意味着可以成为sLPR1 的底物变多,当LPR1 脱落为SLPR1 后,可以通过抑制ERK/MAPK 通路的激活进而促进抑郁症的发生。
综上所述,LPR1 可能会通过介导Aβ和APOE4 的损伤作用,诱导炎症反应并损伤神经系统的正常功能;或通过降低对ADAM10的抑制作用诱导NLRP3炎症小体的产生;也可能通过脱落为sLPR1 加剧炎症反应,并抑制ERK 通路的激活,最终促进抑郁症的发生。
3 结论与展望
LPR1作为一种分布广泛且能与多种配体结合的受体参与了细胞的多种生理功能。但LPR1的异常增加和脱落很可能会导致不良结局的发生。
LRP1异常增加会促进Aβ的内吞,这可能引起细胞内Aβ的异常沉积并导致细胞损伤,同时也意味着sLRP1的底物增多,而sLRP1 参与了神经炎症的发生并能够抑制ERK/MAPK 的激活。此外,LRP1 还介导了APOE4 导致的神经变性。在敲除LPR1或抑制LPR1功能后可以有效抑制炎症反应、Aβ的沉积以及APOE 诱导的神经变性,而神经炎症及损伤都是抑郁症的重要发病机制。因此,LPR1可能作为抑郁症治疗的有效靶点,但仍有必要进行进一步探究。LPR1在机体正常生理功能中有极其重要的作用,维持LPR1 在机体中表达的稳定,抑制LPR1 的脱落和部分下游途径可能有助于预防抑郁症的发生。
猜你喜欢
昆明医科大学学报(2022年1期)2022-02-28
自然杂志(2021年6期)2021-12-23
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年6期)2021-07-31
现代装饰(2018年5期)2018-05-26
哈尔滨医药(2016年3期)2016-12-01
癌变·畸变·突变(2016年3期)2016-02-27
西南医科大学学报(2015年1期)2015-08-22
电源技术(2015年5期)2015-08-22
弹箭与制导学报(2015年1期)2015-03-11
