
Core-level spectroscopy of the photodissociation process of BrCN molecule
2024-01-25 07:11KunZhou周坤andHanWang王涵
Chinese Physics B 2024年1期
Kun Zhou(周坤) and Han Wang(王涵),†
1School of Physical Science and Technology,ShanghaiTech University,Shanghai 201210,China
2Center for Transformative Science,ShanghaiTech University,Shanghai 201210,China
Keywords: x-ray absorption spectroscopy,photodissociation,fewest-switches surface hopping
1.Introduction
Cyanogen halidesXCN (X= F, Cl, Br, I) are a class of molecules that have been extensively studied due to their relevance in atmospheric and combustion chemistry.[1,2]XCN molecules are important precursors for the formation of halogen oxides, which play a crucial role in the depletion of ozone in the stratosphere.[3]The photodissociation ofXCN is a key process in the atmospheric chemistry of halogens and nitrogen, as it can lead to the formation of halogen atoms,which can then react with other atmospheric species, leading to the formation of halogen oxides and other reactive intermediates.[4]Meanwhile, the CN radical is recognized as one of the most significant species in interstellar space,planetary atmospheres,and cometary coma.[5]
XCN,the simplest linear triatomic molecule,has attracted significant attention for its photodissociation dynamics in the A continuum (210 nm–350 nm).[6–15]Two reaction channels have been established through early-stage experimental and theoretical studies[4,16]
Previous studies have determined the major electronic states involved in the photodissociation.[17,18]The parallel transition to3Π0+is the major component of the A absorption band,related to theX∗(2P1/2) channel.The perpendicular transitions to3Π1and1Π1are the minor components, related to theX(2P3/2)channel.[16,19]There is a conical intersection between the potential energy surfaces of3Π0+and1Π1.[7,20]In the ground state, theXCN molecule is linear, while it becomes bent in the excited states.The bending geometry in the excited repulsive states results in high rotational and low vibrational excitation of the CN fragments in bothXandX∗channels.[6,21,22]
The photodissociation ofXCN has been studied using various experimental and theoretical techniques, including laser-induced fluorescence (LIF), velocity map imaging(VMI),ultrafast x-ray spectroscopy andab initiocalculations.LIF is a sensitive technique that can be used to detect the products ofXCN photodissociation, such as halogen atoms and radicals.[23–25]VMI is a powerful technique that can provide detailed information about the velocity distribution and angular distribution of the photofragments.[26]Frankset al.used the brute force orientation method to study the dissociation of BrCN at 230 nm and ICN at 266 nm in a pulsed molecular beam,and found a perpendicular transition component in the absorption process.[27]Wittiget al.demonstrated the effects of the initial quantum state of the parent cyanogen halide molecules on the dynamics of the dissociation process and the vibrational and rotational energy distributions in the fragments, and clearly showed that the photodissociation at 266 nm and 300 K produces CN fragments whose spin is preferentially oriented relative to the rotational angular momentum.[28,29]Fisheret al.studied the photodissociation of ICN and BrCN in the A continuum, and determined the rotational and vibrational distribution of CN in its electronic ground state.[6]Gaoet al.made use of the ion velocity VMI system to study ion-pair dissociations of BrCN,and found an anisotropic distribution of the CN+momentum.[30]Yinet al.studied the photodissociation dynamics of BrCN from 225 nm to 260 nm by time-slice VMI setup,and their findings showed that the internal excitation of CN products in the Br* channel is colder than Br channel, in addition, the photodissociation dynamics at longer wavelengths is found to be different from those at shorter wavelengths in the Br channel.[4]Costenet al.used high-resolution transient frequency modulated absorption spectroscopy to study the nascent Doppler profiles of CN fragments from the A band photodissociation of room temperature ICN.[31]Gaoet al.investigated the dissociative electron attachment process with high-resolution anion VMI apparatus.[26]These studies provide insights into the detection and dynamics ofXCN photodissociation fragments,including the identification of halogen atoms and radicals,as well as the analysis of velocity and angular distributions.Furthermore,investigations into the influence of initial quantum states, rotational and vibrational distributions of CN fragments,and the study of anisotropic properties reveal internal excitations during the dissociation process.However,it remains unclear how the time-dependent changes in electronic states and internal excitations involved in theXCN photodissociation process affect the dynamics and properties of the dissociation products.
X-ray absorption spectroscopy (XAS) is a powerful experimental technique that has been widely used to study the electronic structure and dynamics of molecules.XAS provides valuable information of the electronic and local geometric structure of certain atom, allowing for the investigation of chemical reactions and photodissociation processes.[32]XAS can be employed to investigate the electronic structural rearrangements occurring in various molecular systems during photoinduced bond-breaking reactions by probing the occupied orbitals of organic and organometallic systems.[33,34]Morzanet al.showed that the femtosecond time resolution and atomic specificity of soft-x-ray spectroscopy enable a detailed molecular movie of the ICN photofragmentation dynamics to be captured,including the production of vibrationally hot CN fragments along the I–C dissociation path during the ultrafast relaxation dynamics on the1Π1state.[8]Time-resolved x-ray absorption spectroscopy (TR-XAS) provides a powerful tool for probing the photodissociation ofXCN on a femtosecond timescale.This technique allows for the elucidation of the time-dependent evolution of the electronic structure ofXCN,thereby facilitating the creation of a dynamic molecular movie of the photodissociation process.Consequently, TRXAS holds substantial promise for enhancing our understanding of the photodissociation mechanisms inherent toXCN.
Theoretical calculations provide a complementary approach to experimental studies, offering valuable insights into the electronic structure, potential energy surfaces, reaction pathways,X/X* branching ratio and so on.[4,19]Morokumaet al.theoretically investigated the photodissociation dynamics of ICN based onab initiospin–orbit configuration interaction (SOCI) calculations, including potential energy curves, branching ratio, rotational and vibrational excitation,anisotropy parameter and absorption spectrum.[16,19,35]Bhattacharyyaet al.calculated the structural properties of FCN,ClCN, and their isomers using density functional theory.[36]Baiet al.employedab initiocalculation methods to investigate the low-lying excited states of ClCN.[37]Nathet al.used Fourier grid Hamiltonian (FGH) method based twodimensional mean field methodology to explore the photo dissociation dynamics of the linear triatomic molecule,cyanogen chloride(ClCN)in the polar medium.[38]
In this work,we employ the fewest switches surface hopping theory(FSSH)to study the photodissociation process of BrCN.By analyzing the trajectories from the multi-reference FSSH calculations, we obtain the detailed photodissociation dynamics of BrCN.The x-ray absorption spectroscopy(XAS)of the excited state trajectories is simulated by employing state-averaged complete active-space self-consistent field theory (SA-CASSCF) and RASSI[39]methods.The simulated XAS clearly displays the signatures of conical intersection and the process of molecular fragmentation.
2.Computational methods
Nonadiabatic molecular dynamics simulations for the BrCN molecule were performed with Tully’s fewest switches surface hopping theory[40]as implemented in the SHARC[41,42]software package.The diagonal representation was utilized.This approach involved diagonalizing the electronic Hamiltonian matrix, which is constructed with 10 singlet and 10 triplet spin-free eigenstates in the surface hopping dynamic simulations and 80 singlet and 80 triplet spinfree eigenstates in potential energy surface(PES)simulations,along with spin–orbit interaction, to obtain the spin-mixed,fully adiabatic states with state-averaged complete activespace self-consistent field theory (SA-CASSCF).An active space of 12 electrons in 9 orbitals was used in the excited state calculation with OpenMolcas.[43]ANO-RCC-VDZP basis set was employed and spin–orbit coupling was treated in the atomic mean field (AMFI) approximation.The nuclear motion integration employed the velocity-Verlet algorithm with a time step of 0.1 fs.To account for decoherence, an energy-difference based correction was applied with a parameterα=0.1 Hartree.Since the parallel transition to3Π0+and perpendicular transitions to1Π1are the major components of A continuum, only two electronic initial states for surfacehopping dynamics were considered,specifically exploring the3Π0+and1Π1excited-state PES.For the dynamic simulations,the initial velocities and geometries were sampled from a Wigner distribution centered around the S0minimum geometry.The distribution had an effective temperature of 300 K.The XAS of the excited state trajectories was calculated using SA-CASSCF and RASSI methods, as explained in Ref.[44].30 singlet and 30 triplet states with core holes were included in the XAS calculation.One carbon 1s orbital in the RAS1 active space together with the 9 valence orbitals in the RAS2 space were used in the SA-CASSCF calculation.The XAS in the CK-edge region was obtained by averaging the spectral lineshape over the 125 FSSH trajectories starting from the3Π0+state and 60 trajectories starting from the1Π1state.The spin–orbit natural transition orbitals (SO-NTOs) were calculated with the RASSI module in OpenMolcas.The calculated discrete XAS spectra were convoluted with a Gaussian function with a broadening parameter(σ)of 0.7 eV.Detailed computational methods are explained in supporting information.
3.Results and discussion
3.1.Potential energy surface
We keep the bond length between the C and N atoms in the CN fragment constant at 1.16 ˚A, which enables us to describe the dynamics of the BrCN photodissociation process using a two-dimensional Jacobi coordination system (R,θ).In this coordination system,Rrepresents the distance between the Br atom and the center of mass of the CN fragment,whileθrepresents the angle betweenRand the CN bond.The range ofθis from 0°to 180°,whereθ=0°corresponds to a linear molecular structure of Br–CN,as shown in Fig.1(a).
Photodissociation of BrCN extends the C–Br bond to form a CN· and a neutral Br (2P3/2) or Br∗(2P1/2) atom.As shown in Fig.1(b), potential energy curves are calculated for the bond breaking process.During the C–Br bond breaking process,C–Br bond will extend immediately after the photon excitation.For the PES calculation,the CN fragment is fixed to be the ground state atomic structure of BrCN,and the C–Br bond is elongated from 1.19 ˚A to 3.79 ˚A along the original C–Br bond direction.The3Π0+and1Π1PESs cross when the C–Br distance is 2.57 ˚A(R=3.20 ˚A).

Fig.1.(a) The representation of the BrCN geometry in terms of Jacobi coordinates.(b) The potential energy surface (PES) of BrCN as Br–CN bond increases.
To further study the relationship between PES andθ,the PES of BrCN as the function ofθfor different values ofRis calculated and shown in Fig.2.Asθincreases,the energy of the ground state rises, indicating that the linear Br–C–N geometry is the most stable structure.For all states except the ground state,the lowest energy structure is nonlinear,indicating that during the photodissociation process of BrCN,the CN fragment undergoes rotational movement.This aligns with earlier experimental studies.[4,45]AtR= 2.4 ˚A (Fig.2(a)),all the excited state PES favor bent structures, and there is no crossing between the ground state and excited states asθincreases.However, atR= 3.2 ˚A (Fig.2(b)) and 3.6 ˚A(Fig.2(c)), the excited state potential energy curves become flatter, and there are crossings between the ground state and excited states asθincreases.
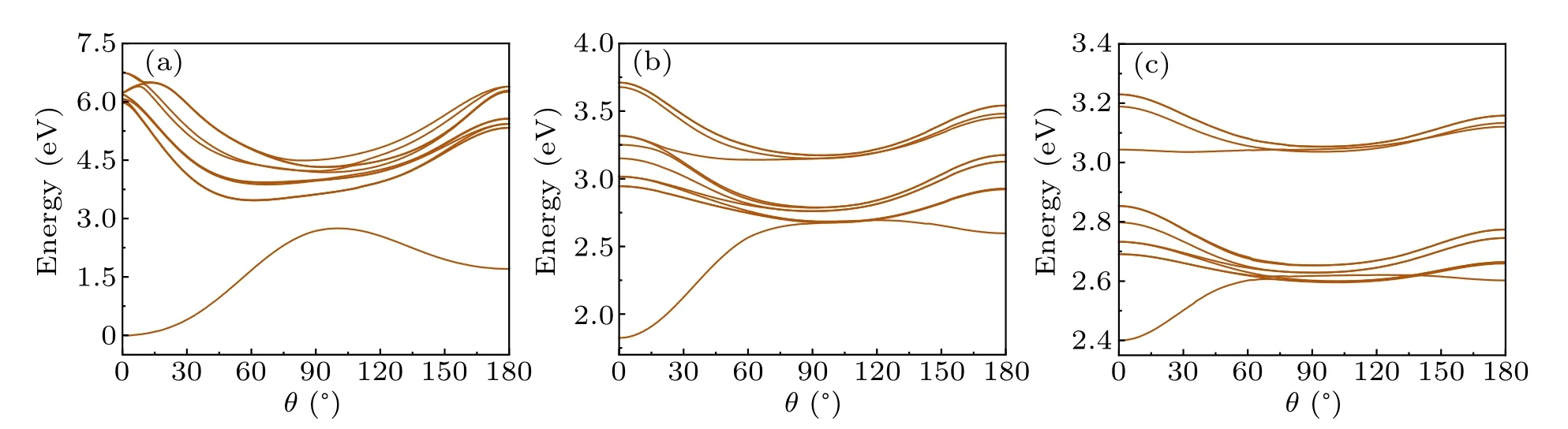
Fig.2.PES of BrCN as bending angle θ changes from 0° to 180°,when R=2.4 ˚A(a),3.2 ˚A(b)and 3.6 ˚A(c),respectively.The ground state energy is set to 0 eV.
3.2.FSSH calculations with SHARC
The evolution of populations in fewest switches surface hopping (FSSH) simulations is investigated by considering states based on the diagonal representation in the SHARC program.This choice is considered natural for FSSH simulations.The evolution of the populations of state 1 to state 12 over the course of the simulation is shown in Fig.3.Figure 3(a) shows the analysis of 125 trajectories starting from3Π0+state,whereas Fig.3(b)displays the analysis of 110 trajectories starting from1Π1state.These results indicate that the photodissociation process of BrCN is in the femtosecond time scale.For the long-time region(50 fs–70 fs),the populations in each state are close,which can be explained by the potential energy surface(PES)in the long C–Br distance region.At the long C–Br distance region,the dissociation channels of BrCN converge into two degenerate states, corresponding to Br (2P3/2) and Br∗(2P1/2).In this case, energy degeneracy occurs between different states, resulting in a close distribution of population among these states.
Since the Br atom is much heavier than the CN fragment,the CN fragment moves rapidly backward right after the photoexcitation,while the bromine atom moves slowly in the opposite direction.During the C–Br bond extending process,depending on the initial Br–C–N angle and the initial velocity of the CN group,the CN group either moves backward straightly or tends to rotate while moving backward.Analyzing the temporal evolution of bond lengths and bond angles in the photodissociation process of BrCN provides insights into the variations in molecular internal structure and dynamics.Figure 4 shows the evolution ofRandθas a function of time during the photodissociation of BrCN with initial excitation states of3Π0+and1Π1.With the increase of time,the distance between Br and CN continues to increase,indicating the photodissociation of BrCN.In Figs.4(c)and 4(d),it can be observed that the angleθundergoes an increase with time,suggesting the rotational motion of the CN fragment during the process of photodissociation.Trajectories starting from3Π0+states exhibit a faster rate of increase in angle with time compared to trajectories starting from1Π1state,indicating that the CN fragment possesses a higher rotational excitation in3Π0+.

Fig.3.Time evolution of the population of the first 12 states in the FSSH calculation, when the initial states are set to (a) 3Π0+ (state 7)and(b) 1Π1 (state 8),respectively.
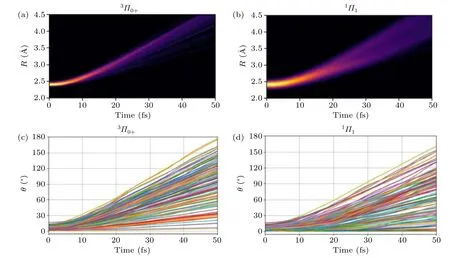
Fig.4.Plots of the change on Jacobi coordinates(R,θ)as a function of time in different initial exited states during the BrCN photodissociation:(a)and(c) 3Π0+ (state 7)and(b)and(d) 1Π1 (state 8).
3.3.Simulated XAS of FSSH trajectories
The XAS simulations of BrCN are performed with Open-Molcas.The Br–C bond dissociation is clearly observed in the XAS simulations as shown in Fig.5.For the trajectories starting from3Π0+state, the XAS exhibits a prominent high energy peak at~286.5 eV and a low energy peak at~285.5 eV in the early stages as a characteristic feature.After about 13 fs,the low energy peak disappears and the high energy peak shifts to higher energy.At the same time,one low-intensity peak appears below the previous two peaks and moves towards the lower energy region.Within 22 fs after photoexcitation, the XAS peaks exhibit pronounced frequency shifts,which correspond to the rotation of the CN fragment and the extension of the Br–C bond.At about 22 fs, the C–Br distance is close to the conical intersection region and the energy of both peaks becomes stable.Regarding the trajectories starting from the1Π1excited state,the character of XAS peaks is more complicated but the shift trending of the XAS peaks is similar to that of the3Π0+state.
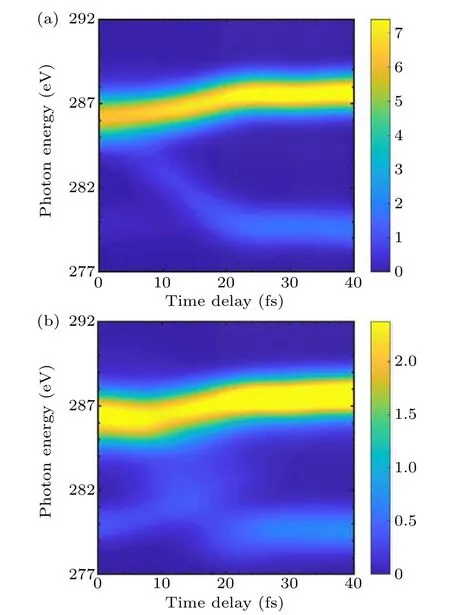
Fig.5.The simulation of carbon K-edge XAS following trajectories starting from (a) 3Π0+ state and (b) 1Π1 state, respectively.The dark blue region means that the absorption is low,while the bright yellow region represents maximal absorption intensity.The transient spectrum is broadened in time by a Gaussian function with a broadening parameter(σ)of 5 fs.
To study the electronic structure dynamics reflected by XAS,Fig.6 shows the XAS of the carbonK-edge of BrCN at different values ofRin the3Π0+state.Each peak is associated with its corresponding natural transition orbitals (NTO).The spin–orbit natural transition orbitals(SO-NTOs)for the XAS peaks are calculated with the RASSI module in OpenMolcas.For the ground state equilibrium structure of BrCN,the XAS of3Π0+state is composed ofπ∗antibonding orbitals in the 285.25 eV–286.85 eV range andσ∗antibonding orbitals at 285.25 eV,as shown in Fig.6(a).As the value ofRincreases,the absorption peak of XAS shifts, indicating that these orbitals are significantly influenced during the photodissociation process.Theσ∗antibonding orbital has axial symmetry along the Br–C bond.When the Br–C bond is elongated,the energy of theσ∗antibonding orbital changes, thereby significantly affecting the carbon 1s toσ∗transition and leading to a pronounced redshift in the absorption peak.In contrast, theπ∗antibonding orbital is mostly localized on the CN fragment,which is less affected by the elongation of the Br–C bond.As a result,the absorption peak only experiences a small blueshift.
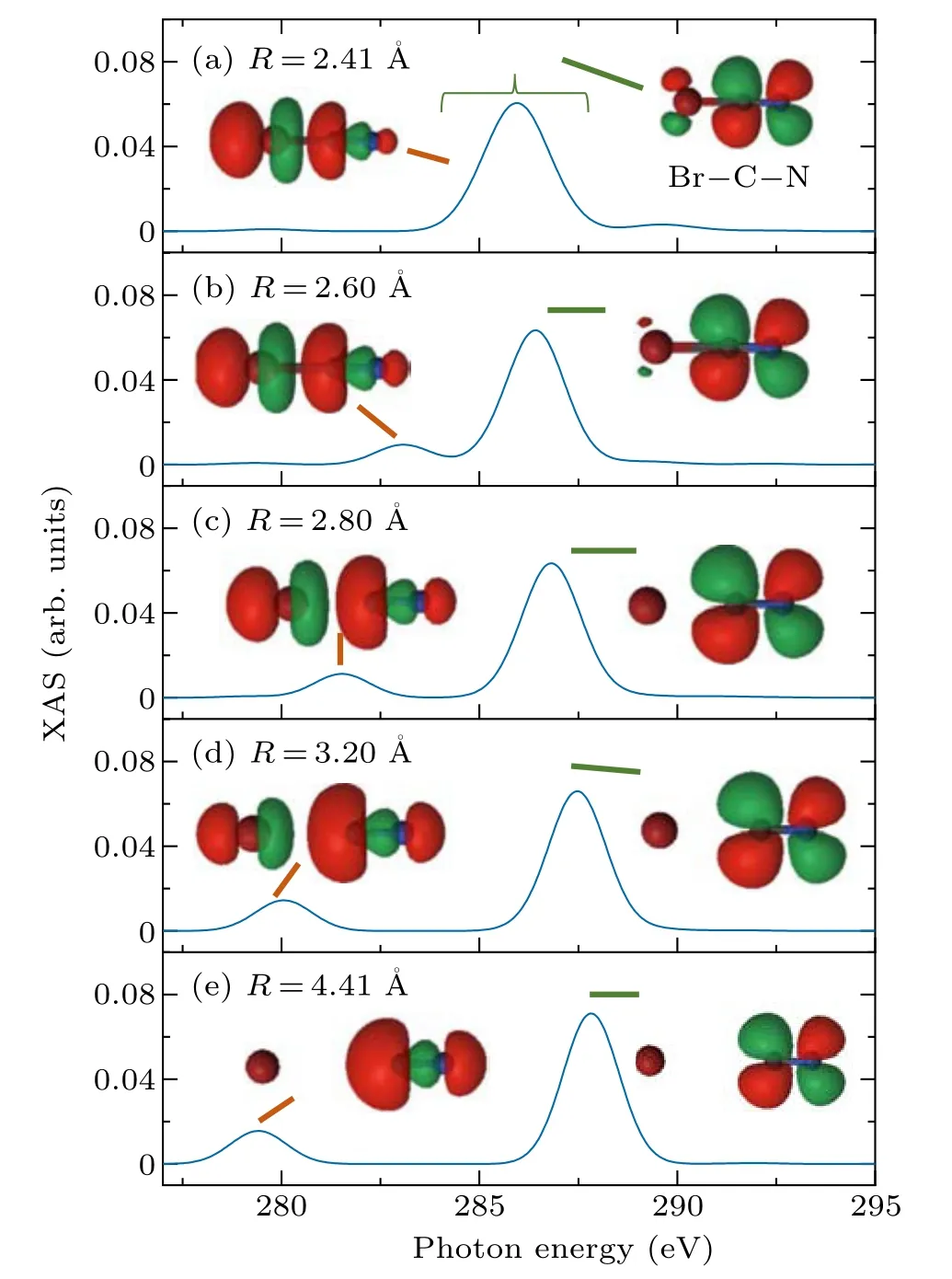
Fig.6.The XAS simulation of the carbon K-edge and corresponding natural transition orbitals (NTO) in the 3Π0+ state were conducted by varying the values of R for BrCN,while(a)R=2.41 ˚A,(b)R=2.60 ˚A,(c)R=2.80 ˚A,(d)R=3.20 ˚A,and(e)R=4.41 ˚A,respectively.
Figure 7 shows the XAS and corresponding NTO in the1Π1state.Compared to the XAS of the3Π0+state, an additional absorption peak at 280.15 eV is observed in XAS atR=2.41 ˚A,this peak is attributed to a transition from the carbon 1s orbital toσbonding orbital,as shown in Fig.7(a).AtR=2.60 ˚A (Fig.7(b)), XAS exhibits an absorption peak at 279.80 eV, corresponding to a transition from the carbon 1s orbital toπbonding orbital.Therefore, the stronger absorption of1Π1in the low-energy region within 0 fs–10 fs in XAS is attributed to the transition from the carbon 1s orbital to the bonding orbital.As the Br–C bond elongates, the absorption peak attributed to the transition from the carbon 1s orbital toσ∗antibonding orbital exhibits a significant redshift, which is caused by the weakening of the Br–C bond.At the same time, the absorption peak associated with the transition from the carbon 1s orbital toπ∗antibonding orbital undergoes a minor blueshift,which observation is consistent with that of the3Π0+state.

Fig.7.The XAS simulation of the carbon K-edge and corresponding natural transition orbitals (NTO) in the 1Π1 state were conducted by varying the values of R for BrCN,while(a)R=2.41 ˚A,(b)R=2.60 ˚A,(c)R=2.80 ˚A,(d)R=3.20 ˚A,and(e)R=4.41 ˚A,respectively.
4.Conclusion
A multi-reference fewest-switches surface hopping(FSSH)simulation is employed to investigate the photodissociation dynamics of cyanogen bromide in the A continuum.Maintaining the linear structure of the BrCN molecule, the3Π0+and1Π1PESs cross when the C–Br distance is 2.57 ˚A.The linear structure of BrCN is the lowest energy structure in the ground state, while all excited states exhibit non-linear structures with the lowest energy,indicating that rotational excitation occurs in the CN fragment during the photodissociation.After being excited from1Σ0+to3Π0+or1Π1state,the distance between the Br and C in BrCN increases rapidly,and the CN fragment undergoes rotation.In trajectories starting from3Π0+,the rotational excitation of the CN fragment is higher than that starting from1Π1.Employing SA-CASSCF and RASSI within OpenMolcas,and excited state trajectories calculated with SHARC, we simulate the XAS of the BrCN photodissociation process.The XAS of carbonK-edge following1Σ0+to3Π0+and1Π1photoexcitation provides detailed dynamical information of the nuclear and electronic dynamics.Peak shifting in the XAS spectra reveals the increase of the Br–C bond length and the rotation of the CN fragment during the photodissociation process of BrCN.
Our research demonstrates that the FSSH method has great potential in predicting the dynamics of bond breaking or photodissociation in small molecules, providing detailed information about the dynamic evolution.Due to the similar linear structure of halogen cyanide molecules,our method can be reasonably extended to the study of photodissociation processes of ICN,FCN,and ClCN.Our method can also be used to investigate other small molecules containing bromine.In the future,we plan to apply our method to investigate the photodissociation processes of other small molecules,such as the photodissociation of ammonia molecule under 200 nm ultraviolet light.By studying the photodissociation dynamics of BrCN,we can better predict the generation and distribution of bromine radicals,leading to improved strategies for mitigating atmospheric pollution and protecting the ozone layer.
Acknowledgments
H.W.and K.Z.were supported by the start-up funding of ShanghaiTech University in China.This work was also supported by a user project at the Molecular Foundry (LBNL)and its computing resources administered by the High-Performance Computing Services Group at LBNL.Work at the Molecular Foundry was supported by the Office of Science and Office of Basic Energy Sciences of the U.S.Department of Energy (Grant No.DE-AC02-05CH11231).This research used resources of the National Energy Research Scientific Computing Center (NERSC), a U.S.Department of Energy Office of Science User Facility located at Lawrence Berkeley National Laboratory(Grant No.DE-AC02-05CH11231).This work was also supported by the High-Performance Computing(HPC)Platform of ShanghaiTech University.We would like to thank Jingxiang Zou for the discussion of NTO analysis.

- Chinese Physics B的其它文章
- Photophysics of metal-organic frameworks: A brief overview
- Anelasticity to plasticity transition in a model two-dimensional amorphous solid
- Ab initio nonadiabatic molecular dynamics study on spin–orbit coupling induced spin dynamics in ferromagnetic metals
- Ultrafast dynamics in photo-excited Mott insulator Sr3Ir2O7 at high pressure
- Universal basis underlying temperature,pressure and size induced dynamical evolution in metallic glass-forming liquids
- Valley filtering and valley-polarized collective modes in bulk graphene monolayers