
Molecular Simulations of Adsorption and Diffusion Behaviors of Benzene Molecules in NaY Zeolite*
2009-05-15 00:25ZHANGZhou张舟LIUHui刘辉ZHUJiqin朱吉钦CHENBiaohua陈标华TIANHuiping田辉平andHEZhenfu贺振富
关键词:刘辉
ZHANG Zhou (张舟), LIU Hui (刘辉),**, ZHU Jiqin (朱吉钦), CHEN Biaohua (陈标华), TIAN Huiping (田辉平) and HE Zhenfu (贺振富)
Molecular Simulations of Adsorption and Diffusion Behaviors of Benzene Molecules in NaY Zeolite*
ZHANG Zhou (张舟)1, LIU Hui (刘辉)1,**, ZHU Jiqin (朱吉钦)1, CHEN Biaohua (陈标华)1, TIAN Huiping (田辉平)2and HE Zhenfu (贺振富)2
1State Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China2Research Institute of Petroleum Processing, SINOPEC, Beijing 100083, China
In the article the Grand Canonical Monte Carlo (GCMC), molecular dynamics (MD), and kinetic Monte Carlo (KMC) simulations with particular focus on ascertaining the loading dependence of benzene diffusion in the zeolite were performed. First, a realistic representation of the structure of the sorbate-sorbent system was obtained based on GCMC simulation. The simulation clearly shows the characteristics of the adsorption sites of the benzene-NaY system, from which two kinds of preferably adsorbing sites for benzene molecules, called SIIand W sites, are identified. The structure thus obtained was then used as a basis for KMC and MD simulations. A comparative study by introducing and comparing two different mechanisms underlying jump diffusion in the zeolite of interest shows that the MS diffusivity values predicted by the KMC and MD methods are fairly close to each other, leading to the conclusion that for benzene diffusion in NaY, the SII→W→SIIjumps of benzene molecules are dominated,while the W→W jumps do not exist in the process. These findings provide further support to our previous conclusionabout the absence of the W→W jumps in the process of benzene diffusion in NaY. Finally, two relations for predicting the self- and MS diffusivities were derived and found to be in fair agreement with the KMC and MD simulations.
benzene, adsorption, diffusion, NaY, Grand Canonical Monte Carlo, kinetic Monte Carlo
1 INTRODUCTION
Zeolites have well defined structures with small pores and have been widely used as catalysts, sorbents and ion exchangers in chemical and petroleum industries [1]. As a catalyst, zeolites can promote many important reactions involving organic molecules,.., cracking, isomerisation and hydrocarbon synthesis. The FAU type zeolites, like the synthetic form NaY investigated in the present study, were reported to have high activity for alkylation reactions of benzene with ethylene or propylene [2]. In these applications, there is a need for accurate description of the diffusive molecular transport through zeolites for a predictive design of the processes. As a result, experimental and theoretical investigations on diffusion of benzene in NaY have received considerable research interest in recent years [2-8].
In a series of modeling and simulation work, Auerbach and coworkers [4, 8-10] studied the jump motion of the guest benzene molecules between the lattice sites in NaY, where there are two types of adsorption sites (donated as SIIsite and W site) for benzene, and the diffusing benzene molecules execute the site-to-site jump motions by assuming the SII→SII, SII→W, W→SII, and W→W jumps. In a proceeding study [11], the kinetic Monte Carlo (KMC) simulations on jump diffusion mechanism of benzene molecules in NaY were reported. Temperature and loading dependences of self-diffusivities of benzene molecules diffusing in NaY were investigated by performing KMC simulations at varying temperatures and loadings. In order to probe the role of W→W jumps of the diffusing benzene molecules in NaY, a comparative study by assuming three diffusion mechanisms was carried out. The results indicated that the diffusing benzene molecules through NaY exhibit three kinds of jumps,.., SII→SII, SII→W and W→SIIjumps. The benzene molecule movements between cages are dominated by SII→W→SIIjumps, while W→W jumps do not occur in the process, which leads to a new loading dependency of self-diffusivity in marked contrast to that derived by Auerbach and coworkers.

2 METHODOLOGY
2.1 Structure and model of NaY zeolite
According to the classification of IZA (International Zeolite Association), NaY belongs to the FAU zeolite. The structure of FAU was reported firstly by Bergerhoff. [13], and then studied by many other researchers [14, 15] upon a basis of X-ray and neutron powder diffraction data.
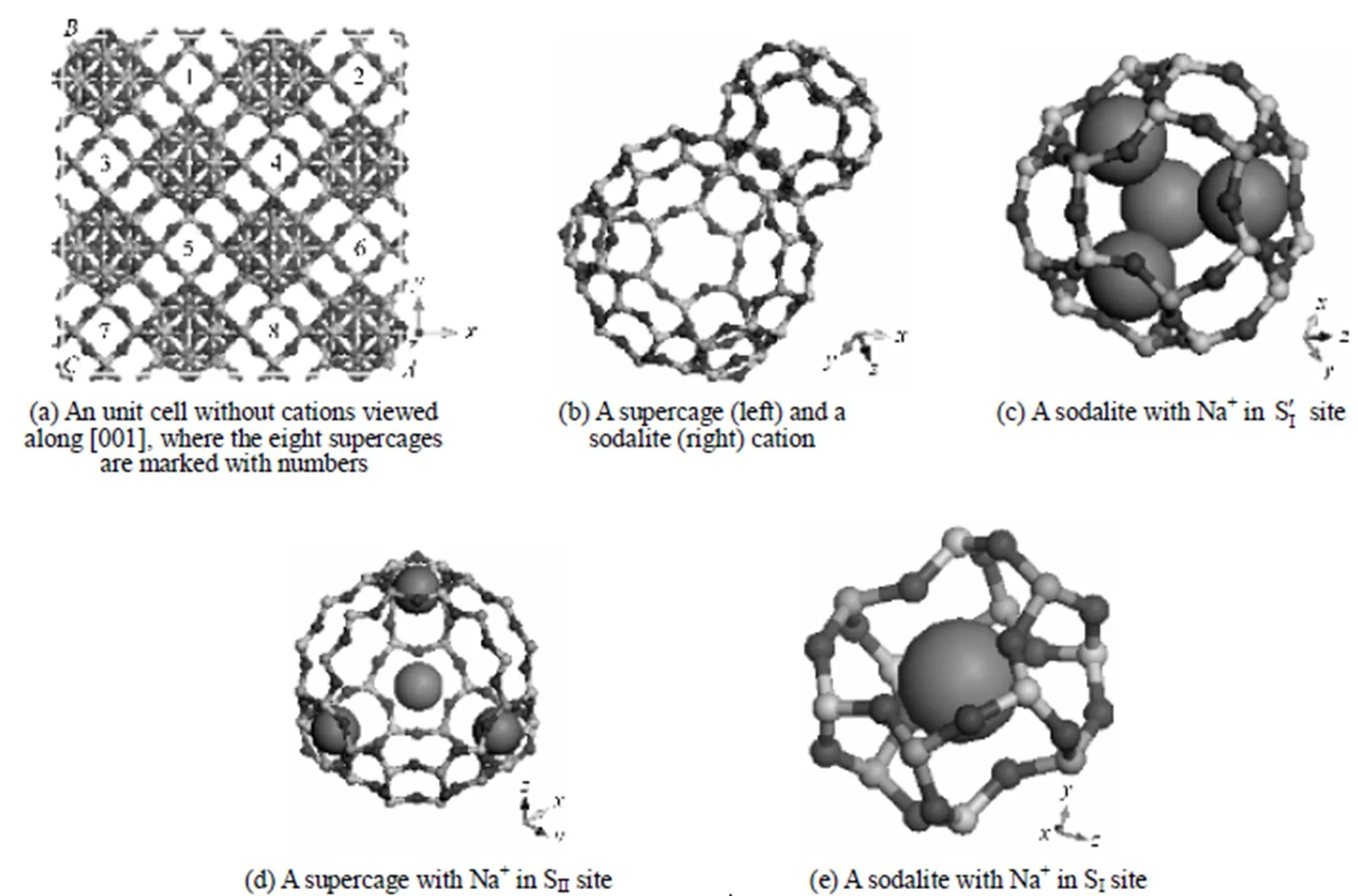
Figure 1 The model for NaY used in MD and GCMC simulations
2.2 Simulation parameters
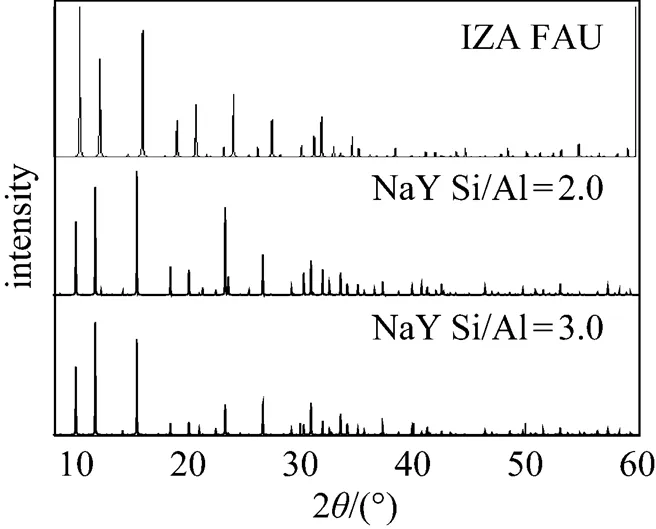
Figure 2 XRD pattern of the NaY model by Reflex, compared with the XRD pattern of FAU from IZA
The total potential energy of the sorbate-sorbent system consists of intra and pair potential energies of zeolite atoms and benzene molecules. The potential energies include both short range and long range interactions. The former part is described by the Lennard- Jones (LJ) potential, and the latter part is described by the Coulombic force, thus
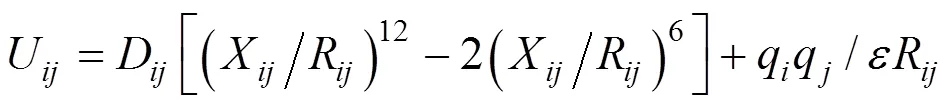
where,andare two different molecules,is the separation distance,andare parameters of the LJ potential,is the partial charge, andis the dielectric constant. The guest molecule is described by an all-atom model. The parameters of the LJ potential and partial charges were obtained from the work of Klein. [3], and are listed in Tables 1 and 2, respectively. The LJ interactions between T atoms and benzene molecules were ignored as they are screened by the oxygen atoms. The LJ interaction was calculated with periodic boundary conditions and a cutoff radius at 1.2014 nm. The Coulombic interaction between the sorbate and sorbent atoms was calculated by Ewald summation. These long-range electrostatic interactions were calculated with all sorbent (including silicon and aluminium) and sorbate atoms.

Table 1 Interatomic LJ potential parameters for interactions

Table 2 Partial charge of atoms ()
2.3 Details of the simulation
2.3.1
In this work, adsorption was computed using grand canonical ensemble () simulations. Classical grand canonical Mote Carlo (GCMC) simulations were carried out at 300 K and 700 K, respectively. The technical details of the method were described in Ref. [17]. In the simulation, 106steps were adopted for equilibration and production. The relative probabilities for attempting these moves were such that 20% of the total number of moves was displacements, 20% rotations, and 60% exchanges with the reservoir (half insertion and half removal).
2.3.2
Molecular dynamics (MD) simulations of benzene diffusion through NaY were performed at four loadings,.., 1, 16, 29, and 40 molecules per unit cell, in theensemble at 700 K using the Discover Module of the Materials Studio Software. Newton’s equations of motion were integrated using the Verlet algorithm with a time step of 1fs. The starting configurations of the zeolite-benzene system were the equilibrated ones obtained from GCMC simulations. The system was allowed to equilibrate for 50 ps (at low loadings) or 100 ps (at high loadings). Then 1.5ns were adopted to collect the trajectories, and the interval of sampling was 150 fs.
Although the MD method provides details of local dynamics of molecular motion, MD simulations are known to be computationally very expensive and limited by numerical and computation capacities. To overcome these limitations, the kinetic Monte Carlo (KMC) method was adopted as a complementary tool to calculate the loading dependency behaviors of benzene diffusion in NaY at 700 K. Details of the KMC methodology adopted in this article have already been described in our previous work [11] and will not be repeated here.
3 RESULTS AND DISCUSSION
3.1 Characteristics of benzene adsorption in NaY
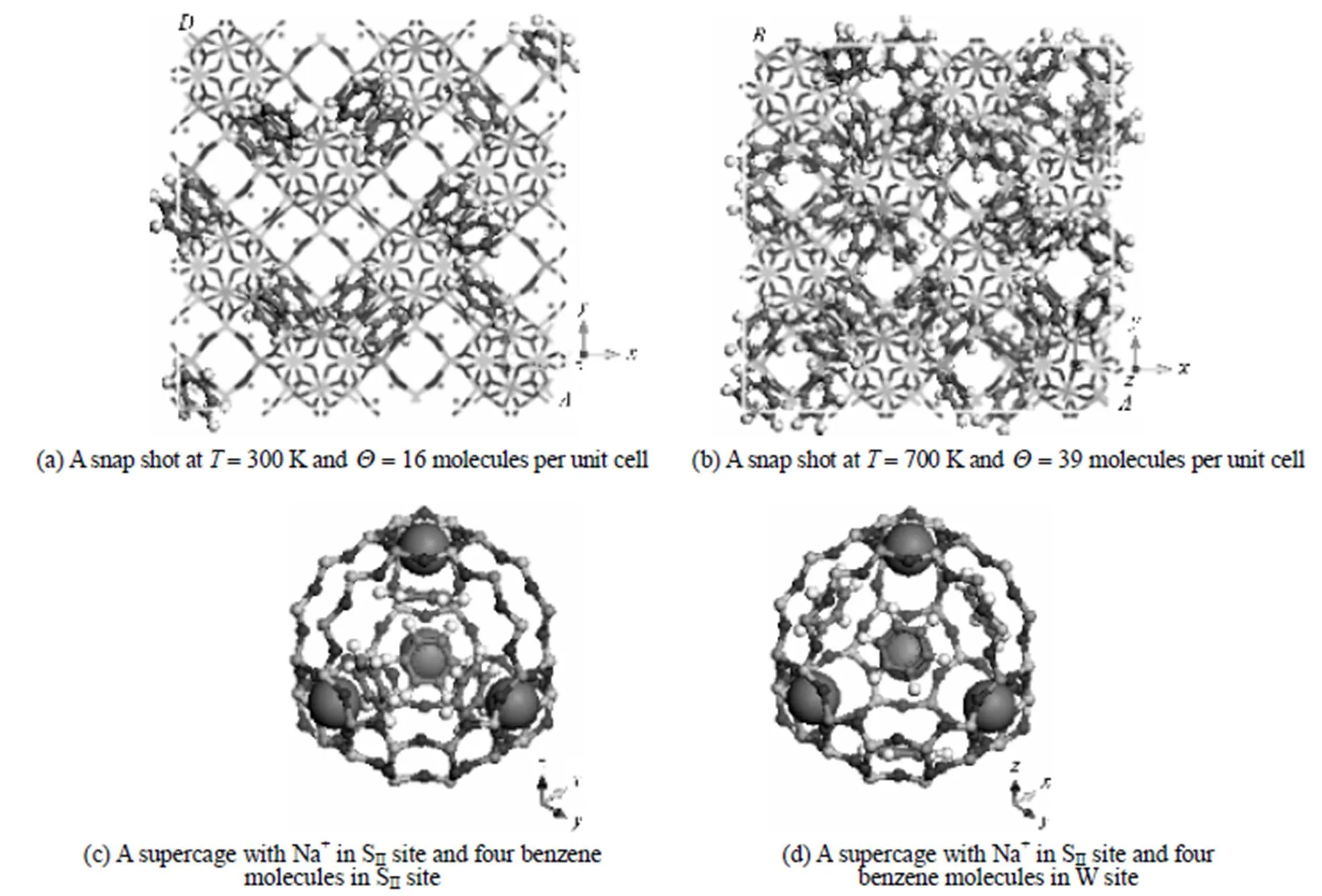

Table 3 Distribution probability of benzene molecules on SII and W sites of NaY (Si/Al2.0) and corresponding binding energies
Based on the simulations, further information on the microscopic adsorption configuration can be obtained as listed in Table 4. This allow us to treat the zeolite framework as a three dimensional lattice solely consisting of the SIIand W sites for the KMC simulations as shown in Fig. 4.

Table 4 The geometrical details of the benzene-NaY adsorption system simulated in the present work and those available in the literature
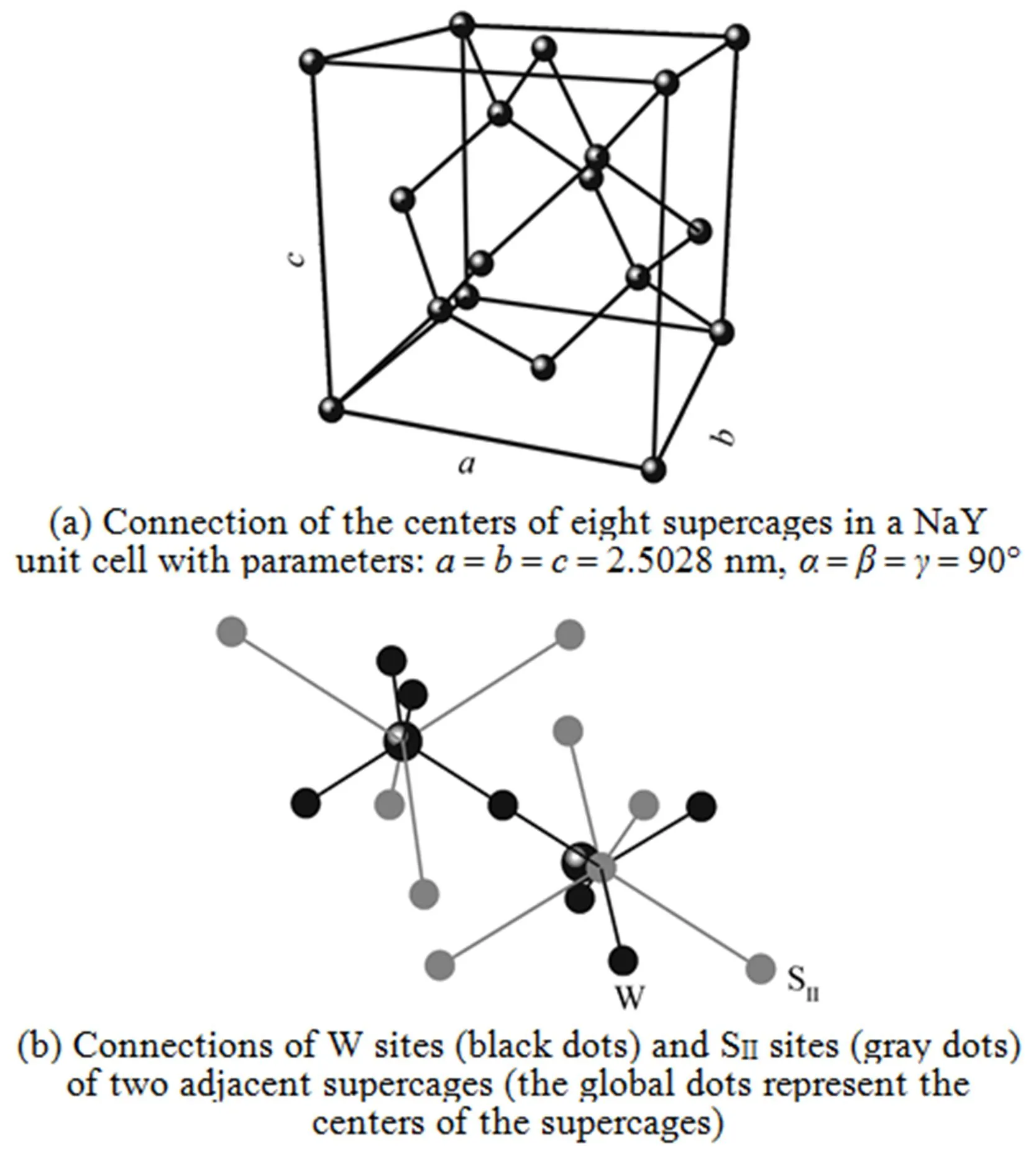
Table 5 Jump frequencies of benzene in NaY (Si/Al2.0) at 700K

3.2 Diffusion behavior of benzene in NaY
The KMC simulation was performed at 700 K (in contrast to 300 K adopted in our previous work [11]), in regard to the loading dependency of MS diffusivity corresponding to the jump mechanisms A and C. The jump frequency values used in the simulation were derived from our previous work [11] and are listed in Table 5. Note that in the mechanism A scenario, a finite Arrhenius prefactor value equal to 4.54×1011s-1for the W→W jumps is assumed to accounted for the W→W jumps and in the mechanism C scenario the W→W jumps are excluded by assuming an zero Arrhenius prefactor value for the W→W jumps. The MD simulation was carried out under the same conditions as those in the KMC simulation, mainly serving as a tool to provide diffusivity data for a direct comparison. The simulated MS diffusivity values by the KMC and MD methods are plotted as a function of molecular loadings as shown in Fig. 5. It is striking that the diffusivity values from the mechanism A show significant discrepancies from the MD data, while the diffusivity values of the mechanism C are in a fair agreement with the MD ones. The two curves departure from each other above a loading of around 1.5. A key ingredient in understanding this phenomenon is the role of the W→W jumps playing in the two diffusion scenarios.

From the comparison and analysis above, it is concluded that the long range motions of benzene are dominated by SII→W→SIIjump, while W→W jump plays no role for benzene diffusion in NaY. This provides the physical ground for building a model to predict the self- and MS diffusivities of benzene in NaY at varying loadings.
The loading dependency of MS diffusivity as described by the following relation [12]was considered for molecular diffusion in MFI zeolite,..,equals to the product of(0), probability of vacant adsorption sites and repulsion interaction between adsorbed molecules.
Different from MFI, there is no channels except supercages interconnected by windows in the topology structure of NaY. Here, in view of the different topology of NaY, the relation is modified,..,equals to the product of(0), probability for a molecule leaving an old supercage, probability of available vacant sites in a new supercage, and repulsion interaction between adsorbed molecules.
In the formulation, three factors are introduced to account for the facts that:
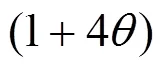
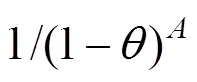
Accordingly, the following relation can be drawn:

whereis described by the dual-site-Langmuir equation

where SIIand W denote the two types of adsorption sites, andis the mole fraction of molecules:
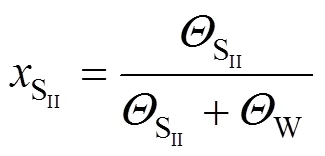

To describe the loading dependence of self- diffusivity, the following relation proposed by Krishna and Paschek is used [19]:
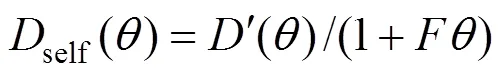
4 CONCLUSIONS
NOMENCLATURE
,,unit cell parameters of zeolite, nm
parameter of LJ potential, kJ·mol-1
′ MS diffusivity, m2·s-1
selfself-diffusivity, m2·s-1

partial charge
distance, nm
potential energy, kJ·mol-1
parameter of LJ potential, nm
mole fraction of molecules
,,unit cell parameters of zeolite, (°)
dielectric constant
loading of benzene in NaY (molecules per supercage)
occupancy of benzene in NaY
Subscripts
,mark of molecules
sat saturated state
SII,W the adsorption sites
1 Krishna, R., “Diffusion of binary mixtures in zeolites: Molecular dynamics simulationsMaxwell-Stefan theory”,, 326, 477-484 (2000).
2 Namuangruk, S., Pantu, P., Limtrakul, J., “Alkylation of benzene with ethylene over faujasite zeolite investigated by the ONIOM method”,, 225, 523-530 (2004).
3 Klein, H., Kirschhock, C., Fuess, H., “Adsorption and diffusion of aromatic hydrocarbons in zeolite Y by molecular mechanics calculation and X-ray powder diffraction”,..., 98, 12345-12360 (1994).
4 Auerbach, S.M., Hensont, N.J., Cheetham, A.K., Metiu, H.I., “Transport theory for cationic zeolites: Diffusion of benzene in Na-Y”,..., 99, 10600-10608 (1995).
5 Bremard, C., “Organometallic chemistry of group VI metals in the void space of zeolites”, Coordination Chemistry Reviews, 178-180, 1647-1677 (1998).
6 Auerbach, S.M., Metiu, H.I., “Diffusion in zeolitescage-to-cage kinetics: Modeling benzene diffusion in Na-Y”,..., 105, 3753-3760 (1996).
7 Paschek, D., Krishna, R., “Monte Carlo simulations of self- and transport-diffusivities of 2-methylhexane in silicalite”,...., 2, 2389-2394 (2000).
8 Saravanan, C., Auerbacha, S. M., “Modeling the loading dependence of diffusion in zeolites (I) Analytical theory for benzene in Na-Y”,..., 107 (19), 8120-8131 (1997).
9 Saravanan, C., Auerbacha, S. M., “Modeling the loading dependence of diffusion in zeolites (II) Kinetic Monte Carlo simulations of benzene in Na-Y”,..., 107 (19), 8133-8137 (1997).
10 Auerbach, S.M., Horia, I.M., “Diffusion in zeolitescage-to-cage kinetics: Modeling benzene diffusion in Na-Y”,..., 105 (9), 3753-3760 (1996).
11 Zhang, Z., Liu, H., Chen, B.H., “Self-diffusivity of benzene in zeolite Y: Different jump diffusion mechanisms investigated by KMC simulations”,.... (), 57 (5), 1147-1152 (2006). (in Chinese)
12 Zhang, Z., Liu, H., Chen, B.H., “Occupancy dependence of M-S diffusivity of single component in MFI zeolite”,.... (), 56 (11), 2054-2058 (2005). (in Chinese)
13 Bergerhoff, G., Baur, W. H., Nowacki, W., “The crystal structure of faujasite”,,, 193-200 (1958).
14 Olson, D.H., “Crystal structure of the zeolite nickel faujasite”,..., 72, 4366-4373 (1968).
15 Fitch, A.N., Jobic, H., Renouprez, A., “Localization of benzene in sodium-Y zeolite by powder neutron diffraction”,..., 90, 1311-1318 (1986).
16 Klein, H., Kirschhock, C., Fuess, H., “Adsorption and diffusion of aromatic hydrocarbons in zeolite Y by molecular mechanics calculation and X-ray powder diffraction”,..., 98, 12345-12360 (1994).
17 Frenkel, S., Understanding Molecular Simulation—From Algorithms to Applications, Academic Press, Amsterdam (1996).
18 Klein, H., Fuess, H., Schrimpf, G., “Mobility of aromatic molecules in zeolite NaY by molecular dynamics simulation”,..., 100, 11101-11112 (1996).
19 Krishna, R., Paschek, D., “Verification of the Maxwell-Stefan theory for tracer diffusion in zeolites”,..., 85, 7-15 (2002).
2008-11-20,
2009-06-04.
the State Key Development Program for Basic Research of China (2004CB719505), and the National Natural Science Foundation of China (20625621).
** To whom correspondence should be addressed. E-mail: hliu@mail.buct.edu.cn
猜你喜欢
四川生理科学杂志(2022年4期)2022-12-06
Chinese Physics B(2022年8期)2022-08-31
江苏教育·书法教育(2021年10期)2021-11-22
作文新天地(小学版)(2021年4期)2021-09-09
Plasma Science and Technology(2020年9期)2020-12-20
歌剧(2017年11期)2018-01-23
艺术评论(2017年8期)2017-10-16
中学生数理化·高一版(2017年2期)2017-04-25
喜剧世界(2016年1期)2016-12-06
意林(2015年20期)2015-10-21
 Chinese Journal of Chemical Engineering2009年4期
Chinese Journal of Chemical Engineering2009年4期
- Chinese Journal of Chemical Engineering的其它文章
- A Review of Techniques for the Process Intensification of Fluidized Bed Reactors
- Removal of Uranium (VI) by Fixed Bed Ion-exchange Column Using Natural Zeolite Coated with Manganese Oxide*
- Challenges in Study of Single Particles and Particle Swarms*
- Influences of the [Co2+]/[Co3+] Ratio on the Process of Liquid-phase Oxidation of Toluene by Air*
- A Comparative Study of the Performance of Symmetric and Asymmetric Mixed-conducting Membranes*
- Development on the Technique of Total Recovery of Benzoic Acid Residue