
具有π共轭骨架的Salen-卟啉型同、异双核配合物的合成及谱学性质
2012-03-06 04:43王官耀闫伟伟张晓红阮文娟朱志昂
物理化学学报 2012年12期
王官耀 闫伟伟 张晓红 阮文娟 朱志昂
(南开大学化学学院,天津300071)
1 引言
配位化学是目前最活跃的学科之一,近年来,随着配位化学研究的深入,具有多样化结构的多核金属配合物的研究日趋受到重视.1-5以金属配合物为基础的各种功能材料发展十分迅速,对多核金属配合物性质的研究可以为开发制备新型的光、电、磁等功能材料提供实验和理论指导.卟啉及其金属配合物作为一类特殊的大环共轭体系,具有优越的物理、化学及光学特征,在光电材料、分子识别、分子组装、有机合成、光谱分析等领域有广阔的应用前景.6-12而包含另一类常见的配体Salen(N, Nʹ-bis-(saliylaldehyde)ethylendiamine)或其衍生物的配位化合物则主要应用于不对称催化领域,13-16对Salen型配合物的光、电、磁等各项功能性质的探讨近些年来也逐渐成为热点.17-21既然卟啉类化合物与Salen型配合物均能与一系列的过渡金属结合形成稳定的化合物,由此可以构建具有特殊光、电、磁等性质的Salen-卟啉型双核金属配合物,不仅对开发新型功能材料具有重要意义,而且在光学器件、分子开关、电极修饰及磁性材料等领域也具有重要的潜在应用价值.1,22,23
本文合成了文献中未见报道的多种具有π共轭骨架的新型Salen-卟啉型金属配合物,采用1H NMR、紫外-可见吸收光谱、红外光谱和荧光光谱等多种表征手段对它们进行了研究.
2 实验部分
2.1 仪器和试剂
Mercury Vx 300 MHz核磁共振仪(美国Varian公司生产),氘代三氯甲烷(CDCl3)为溶剂,四甲基硅烷(TMS)为内标;MAGNA-560傅里叶变换红外光谱仪(美国Nicolet公司生产);TRACE DSQ型质谱仪(美国Thermofinnigan公司生产);Shimadzu UV-2450紫外-可见分光光度(日本Shimadzu公司生产); Shimadzu RF-5301 PC型荧光光谱仪(日本Shimadzu公司生产).
吡咯、苯甲醛、三乙胺、三氯甲烷均为分析纯试剂,使用前按试剂手册24方法处理.碳酸钾(分析纯)于马弗炉中400°C焙烧2 h待用,其余试剂也均为分析纯,可直接使用.
2.2 合 成
合成路线见示意图1、2和3.
5,10,15,20-四苯基-6ʹ,7ʹ-二氨基喹啉[2,3-bʹ]卟啉(化合物A)按照文献25-28合成,化合物1-8按如下方法合成.
2.2.1 化合物1和2的合成
将3,5-二叔丁基-2-羟基苯甲醛250 mg(1 mmol)溶于10 mL无水甲醇中,加入适量无水碳酸钾,搅拌下滴加化合物A(80 mg)的三氯甲烷溶液.混合溶液在室温下避光搅拌3天.过滤,蒸除溶剂,以二氯甲烷/石油醚为1:3(体积比,下同)的混合液作为淋洗剂,用硅胶柱分离提纯,收集第一色带,产率为10%;再以二氯甲烷/石油醚为2:1的混合液为淋洗剂,收集第二色带,产率为64%.其中第一色带为化合物2,第二色带为化合物1.
化合物 1(C65H54N8O):1H NMR(CDCl3,300 MHz),δ-2.56(s,2H,pyrrole N―H),1.41(s,18H, t-Bu―H),4.54(s,2H,―NH2),7.02(s,1H,quinoxaline H),7.35(s,1H,quinoxaline H),7.38(d,J=2.326 Hz,1H,ArO―H),7.57(d,J=2.146 Hz,1H,ArO―H), 7.76-7.79(m,12H,aryl H),8.17(d,J=1.557 Hz,4H, aryl H),8.21-8.24(m,4H,aryl H),8.71(s,2H,β-pyrrolic H),8.77(s,1H,N=CH),8.92-8.93(m,4H, β-pyrrolic H),13.03(s,1H,O―H).MS,m/z 963.64 (M+H)+.
化合物2(C80H74N8O2):1H NMR(CDCl3,300 MHz),δ-2.56(s,2H,pyrrole N―H),1.40(s,18H, t-Bu―H),1.50(s,18H,t-Bu―H),7.35(d,J=7.8 Hz, 2H,ArO―H),7.52(d,J=2.403 Hz,2H,ArO―H), 7.55(s,2H,quinoxaline H),7.79-7.84(m,12H,aryl H),8.19(d,J=7.00 Hz,4H,aryl H),8.22-8.25(m,4H, aryl H),8.72(s,2H,β-pyrrolic H),8.77(s,2H,N=CH),8.96(m,4H,β-pyrrolic H).13.36(s,2H,O―H). MS,m/z 1179.81(M+H)+.
2.2.2 模板法合成化合物3-6
(1)将20 mg化合物1溶于4 mL二氯甲烷中,加入醋酸镍的甲醇饱和溶液8 mL,避光条件下回流20 min,加入36 mg 3,5-二叔丁基-2-羟基苯甲醛,30 min后有深紫红色沉淀出现,继续反应直至沉淀的量不再变化.过滤,将沉淀风干后,用适量二氯甲烷溶解,并用蒸馏水洗涤至水相呈中性.用无水硫酸镁干燥,蒸除溶剂.用硅胶柱分离提纯,以二氯甲烷/石油醚为1:2的混合液为淋洗剂,收集第一色带,产率为30%;收集第二色带,产率为45%.二者均为紫红色固体,其中第一色带为化合物4,第二色带为化合物3.

示意图1 化合物1和2的合成路线Scheme 1 Synthesis route of compounds 1 and 2
化合物3(C80H72N8NiO2):1H NMR(CDCl3,300 MHz),δ-2.59(s,2H,pyrrole N―H),1.42(s,18H, t-Bu―H),1.49(s,18H,t-Bu―H),7.48(d,J=2.371, 2H,quinoxaline H),7.52(s,2H,ArO―H),7.55(s,2H, ArO―H),7.77-7.79(m,4H,aryl H),7.86(s,2H,aryl H),7.88(s,2H,aryl H),7.96-7.98(m,4H,aryl H), 8.16-8.21(m,4H,aryl H),8.24(m,4H,aryl H),8.40 (s,2H,N=CH),8.70(s,2H,β-pyrrolic H),8.93(s, 4H,β-pyrrolic H).MS,m/z 1235.85(M+H)+.

示意图2 化合物3-6的合成路线Scheme 2 Synthesis route of compounds 3-6

示意图3 化合物7和8的合成路线Scheme 3 Synthesis route of compounds 7 and 8
化合物4(C80H70N8Ni2O2):1H NMR(CDCl3,300 MHz),δ 1.39(s,18H,t-Bu―H),1.48(s,18H,t-Bu―H),7.18(s,2H,quinoxaline H),7.35(d,J=7.8 Hz,2H, ArO―H),7.47(d,J=2.450 Hz,2H,ArO―H),7.92(s, 4H,aryl H),7.75-7.78(m,4H,aryl H),7.89-7.90(m, 8H,aryl H),7.98(m,4H,aryl H),8.34(s,2H,N=CH),8.63(s,2H,β-pyrrolic H),8.69(s,4H,β-pyrrolic H).MS,m/z 1291.64(M+H)+.
(2)将20 mg化合物1溶于4 mL二氯甲烷中,加入醋酸镍的甲醇饱和溶液8 mL,避光条件下回流20 min,加入20 μL水杨醛,45 min后有深紫红色沉淀出现,继续反应直至沉淀的量不再变化为止.过滤,将沉淀风干后,用适量二氯甲烷使之溶解,蒸馏水洗涤至水相呈中性.用无水硫酸镁干燥,蒸除溶剂.用硅胶柱分离提纯,以二氯甲烷/石油醚为1:2的混合液为淋洗剂,收集第一色带,产率为69%;再以二氯甲烷/石油醚为2:1的混合液为淋洗剂,收集第二色带,产率为29%,蒸除溶剂得紫红色固体.二者均为紫红色固体,第一色带为化合物6,第二色带为化合物5.
化合物5(C72H56N8NiO2):1H NMR(CDCl3,300 MHz),δ-3.44(s,2H,pyrrole N―H),1.46(m,9H, t-Bu―H),1.52(m,9H,t-Bu―H),6.73(s,1H,quinoxaline H),6.99(s,1H,quinoxaline H),7.04(s,1H,ArO―H),7.42(s,2H,ArO―H),7.60(s,2H,ArO―H),7.66 (s,1H,ArO― H),7.72-7.77(m,4H,aryl H), 7.80-7.85(m,6H,aryl H),7.88(m,4H,aryl H),7.99 (s,6H,aryl H),8.35(s,1H,N=CH),8.47(s,1H,N=CH),8.57(s,2H,β-pyrrolic H),8.75-8.88(m,4H, β-pyrrolic H).MS,m/z 1123.67(M+H)+.
化合物6(C72H54N8Ni2O2):1H NMR(CDCl3,300 MHz)δ 1.40(s,9H,t-Bu-H),1.48(s,9H,t-Bu―H), 7.04(s,1H,quinoxaline H),7.15(s,1H,quinoxaline H),7.32(s,1H,ArO―H),7.39(s,1H,ArO―H),7.42 (s,1H,ArO―H),7.45(s,1H,ArO―H),7.52(s,1H, ArO―H),7.55(s,1H,ArO―H),7.73-7.76(m,10H, aryl H),7.84-7.86(m,6H,aryl H),7.92-7.94(m,4H, aryl H),8.15(s,1H,N=CH),8.23(s,1H,N=CH), 8.54(s,2H,β-pyrrolic H),8.64-8.69(m,4H,β-pyrrolic H).MS,m/z 1179.302(M+H)+.
2.2.3 化合物7和8的合成
将10 mg化合物3(或5)溶于15 mL二氯甲烷,加入醋酸锌的甲醇饱和溶液4 mL,避光室温搅拌,直至TLC显示反应完成.反应混合物用蒸馏水洗涤三次后,用无水硫酸镁干燥,蒸除溶剂.以二氯甲烷/石油醚为2:1的混合液为淋洗剂,用硅胶柱分离提纯,收集主色带,得化合物7(或8).
化合物7(C80H70N8NiZnO2):1H NMR(CDCl3,300 MHz)δ 1.44(s,18H,t-Bu―H),1.50(s,18H,t-Bu―H),7.24(s,2H,quinoxaline H),7.33(d,J=4.446 Hz, 2H,ArO―H),7.49(d,J=2.122 Hz,2H,ArO―H), 7.79-7.83(m,4H,aryl H),7.93-7.94(m,4H,aryl H), 8.01-8.02(m,8H,aryl H),8.16-8.17(m,4H,aryl H), 8.34(s,2H,N=CH),8.62(s,2H,β-pyrrolic H), 8.83-8.89(m,4H,β-pyrrolic H).MS,m/z 1296.40(M+ H)+.
化合物8(C72H54N8NiZnO2):1H NMR(CDCl3,300 MHz)δ 1.49(m,9H,t-Bu―H),1.54(m,9H,t-Bu―H),6.51(s,1H,quinoxaline H),6.75(s,1H,quinoxaline H),6.84(s,1H,ArO―H),7.22(s,2H,ArO―H), 7.53(s,2H,ArO― H),7.61(s,1H,ArO― H), 7.75-7.79(m,4H,aryl H),7.87-7.91(m,6H,aryl H), 7.93-8.14(m,10H,aryl H),8.31(s,1H,N=CH),8.42 (s,1H,N=CH),8.50(s,2H,β-pyrrolic H),8.64-8.76 (m,4H,β-pyrrolic H).MS,m/z 1185.31(M+H)+.
3 结果与讨论
3.1 核磁共振氢谱
在化合物1中,卟啉环内的吡咯氢的化学位移在-2.56处,侧链―NH2、CH=N和―OH的质子峰化学位移分别在4.54、8.77和13.03处.在化合物A中,―NH2的质子峰化学位移处于3.96处.可知由化合物A生成化合物1,―NH2的质子峰向低场移动,这是因为化合物1中―NH2与一侧的―OH之间形成了氢键,H质子受到了很大的去屏蔽效应.29这也说明化合物A中的一个―NH2与醛基发生反应,生成化合物1,质谱结果进一步验证了这个结论.
在化合物2中,卟啉环内的吡咯氢的化学位移在-2.56处,侧链中CH=N和―OH的质子峰化学位移分别在8.77和13.36处,并且氢质子的个数都是两个,没有出现化合物A中3.96处附近的―NH2的质子峰,在化合物1中出现的4.54处的―NH2的质子峰也同样消失,说明化合物A中2个―NH2与2个醛基发生反应,生成化合物2,质谱结果同样验证了这个结论.
在自由卟啉化合物中,由于卟啉环流效应在环内产生一个对抗外加磁场的感应磁场,受其影响,卟啉环内的H原子处于卟啉环共轭结构的屏蔽区,使其质子的化学位移移向高场,化学位移值小于零.以金属Ni2+离子为模板,分别与化合物1中存在的胺基及两种水杨醛衍生物发生配位反应,生成两类不同的配合物.
在第一类产物3和5中,卟啉环内的H质子峰依然存在,化学位移分别在-2.59和-3.44处;而―OH质子峰则消失.这说明金属Ni2+离子只与化合物1和相应的水杨醛反应所形成的Salen型席夫碱配体配位,并未嵌入卟啉环内的空穴中,没有形成金属卟啉,即生成单核Salen-卟啉型镍金属配合物.相应质谱图中,(M+H)+正离子峰分别为1235.85、1123.67,与其理论值相符合.在第二类产物4和6中,卟啉环内的H质子峰消失,说明了金属卟啉配合物的形成;而―OH质子峰的消失,同样说明Salen型金属配合物的形成.以上结论证实双核Salen-卟啉型镍配合物的生成,所对应的质谱(M+H)+正离子峰分别为1291.64和1179.30,与其理论值相符合.
在化合物3和5与金属Zn2+离子发生配位生成的产物中,原分别位于-2.59处和-3.44处的卟啉环中的H质子峰消失,说明金属Zn2+与卟啉环配位,即形成了对称型异双核Salen-卟啉型配合物7和不对称型异双核Salen-卟啉型配合物8,质谱结果进一步验证了该结论.
对比金属配位前后的核磁谱图不难发现,受中心金属离子亲核作用的影响,金属配合物周围电子云密度降低,磁屏蔽作用减小,质子峰的化学位移均有向高场移动的趋势,并且在双核金属配合物(化合物4、6和7)中此现象更为明显.化合物3是化合物2的单核金属配合物,而化合物4和7则是化合物2的双核金属配合物.不难发现,在化合物2中, CH=N的质子峰的化学位移在8.77处,而在化合物3、4和7中,则分别移到了8.40、8.34和8.34处.化合物6相当于金属Ni2+离子与化合物5的卟啉环配位而成.在化合物5中,CH=N的质子峰的化学位移分别处于8.35和8.47;在化合物6中则分别移到了8.15和8.23.由此可知,卟啉环中心配位的金属离子对电子云密度的影响不局限于卟啉环,由于整个分子具有π共轭骨架,其对处于卟啉环一侧的Salen型配体也产生了较大的影响.
3.2 红外光谱
红外光谱采用石蜡糊法,在4500-400 cm-1范围内摄谱,主要红外吸收谱带的归属列于表1.
化合物1在3347、965、731 cm-1处出现N―H伸缩振动吸收峰,而在化合物4和6(通过模板法得到)的红外光谱中,这三条特征谱带消失,同时在1009和1007 cm-1处分别出现一个新的强伸缩振动峰,该峰归属于Ni―N键引起的卟啉环的振动,30这说明卟啉环上的N―H两个质子被金属Ni2+离子取代而形成稳定的金属卟啉配合物.另外化合物4和6分别在1618和1613 cm-1出现的强C=N伸缩振动峰,对比化合物1处于1610 cm-1处的强C=N伸缩振动峰,可知金属配合物的振动吸收峰向低波数方向移动,说明金属Ni2+离子与偶氮甲烷基的N原子之间形成了Ni―N键.31伴随―OH伸缩振动峰的消失,在550和420 cm-1左右出现的两个峰分别归属于Ni―O键和Ni―N键的特征振动峰.这说明了化合物4和6中含有席夫碱结构,证明了化合物4和6是双核镍Salen-卟啉型金属配合物的结论.
化合物3和5的N―H伸缩振动特征吸收峰的三条谱带依旧存在,这说明两者的卟啉环内均无金属离子与吡咯氮原子配位.在1615 cm-1左右均出现强C=N伸缩振动峰,而―OH伸缩振动峰消失的同时,出现550和420 cm-1左右的Ni―O键和Ni―N键的特征振动峰,则说明化合物3和5存在席夫碱结构,证明了化合物3和5是单核Salen-卟啉型镍金属配合物的结论.
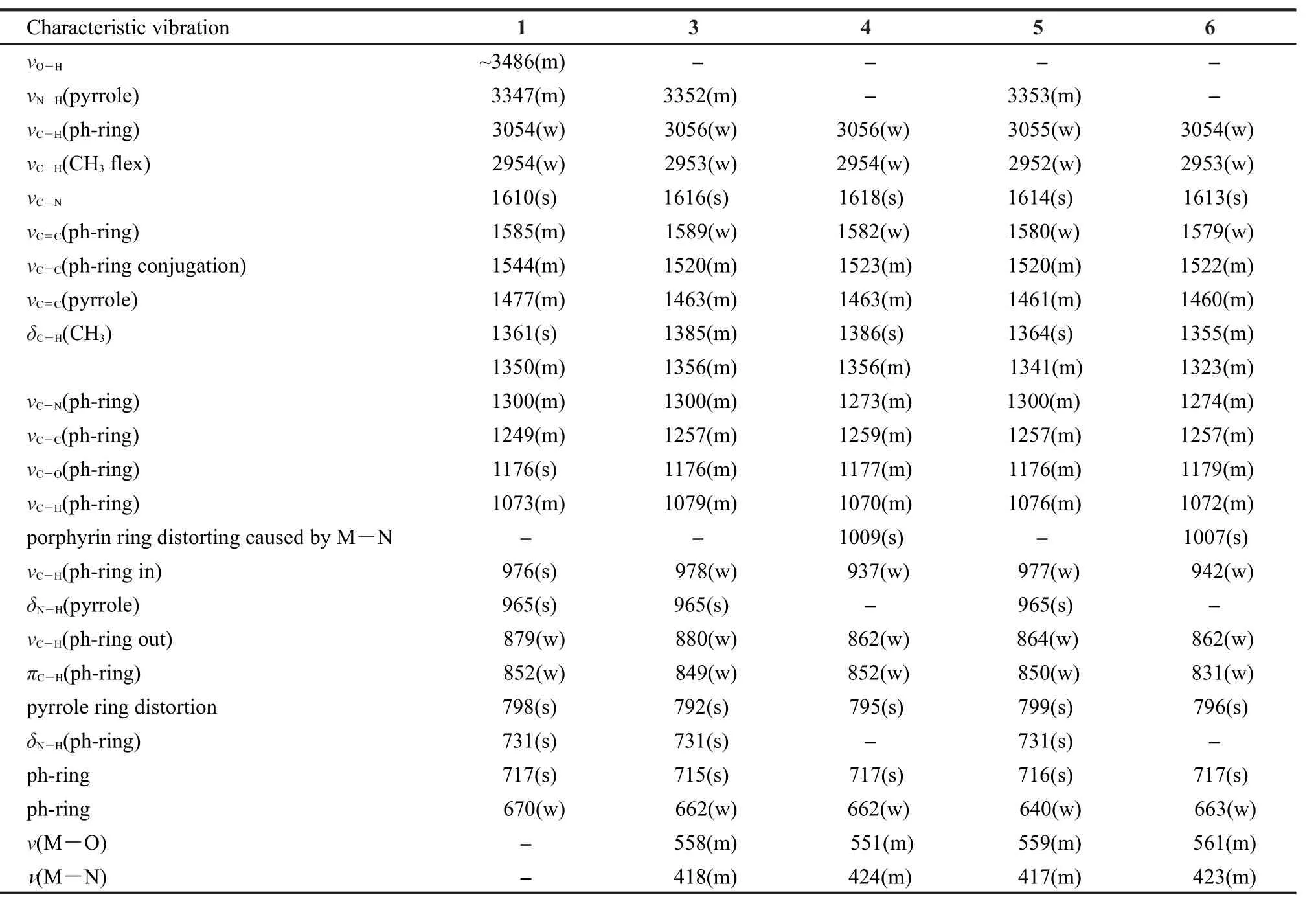
表1 化合物1、3、4、5、6的FTIR特征振动值(单位:cm-1)Table 1 FTIR characteristic vibration values(in cm-1)of compounds 1,3,4,5,6
3.3 紫外-可见光谱
化合物1-8的紫外-可见光谱数据列于表2.
卟啉类化合物在可见光区的电子吸收是由卟啉环大π键共轭体系的π-π*跃迁所产生的.卟啉一般具有特殊的紫外-可见吸收,自由卟啉具有一个较强的Soret带和四个Q带.当卟啉环内吡咯氮原子与金属离子配位后,Soret带将会发生移动,Q带也会发生变化,最显著的就是Q带数目的减少.32
化合物2的吸收光谱在440 nm附近出现的强吸收带为Soret带,由卟啉部分a1u(π)-eg(π*)的跃迁而产生.在528、564、600、651 nm处出现的四个较弱吸收带为Q带,由卟啉的a2u(π)-eg(π*)的跃迁而产生.33,34卟啉部分与金属离子配位形成金属配合物后,卟啉环的对称性增强,Q带数目减少.对比化合物2的Soret带与Q带,金属离子取代卟啉环内的两个氢原子后,Soret带发生了蓝移,Q带数目减少为两个.35
Salen型化合物的紫外-可见光谱的特征峰一般可归为这样几类,处于250-280 nm区域的吸收峰为苯环的π-π*跃迁所产生;在300-320 nm区域的吸收峰可归属于偶氮甲烷基生色团的π-π*跃迁所引起;而金属配合物一般在400-500 nm的吸收谱带则是由于金属与Salen型配体的配位原子N2O2间的d-π*电荷转移跃迁所产生;600 nm以上部分有时可观察到d-d跃迁所致的弱电子吸收峰.Salen型金属配合物的电子吸收谱带发生红移或蓝移与金属离子的电负性及金属本身内在的性质有关.根据现代分子轨道理论,很多因素都可以影响整个共轭体系的电子云密度,从而使吸收峰发生不同程度的红移或蓝移.36-42
本文设计合成的Salen-卟啉型化合物,由于其具有π共轭骨架,卟啉和Salen的特征峰与卟啉和Salen单体化合物相比会有一些变化.分析图1中的曲线及表2中的数据可以看出,化合物1-8的λ1、λ2和λ4三个吸收带可归于Salen部分;化合物1、2的λ3和λ5、λ6、λ7、λ8吸收带可分别归于卟啉部分的Soret带和四个Q带;化合物3-8的λ3和λ5、λ6吸收带可分别归于卟啉部分的Soret带和两个Q带,化合物3、5、6、7、8的λ8吸收带的弱电子吸收峰则可能是由于d-d跃迁所致及卟啉与Salen形成的共轭结构所产生的.
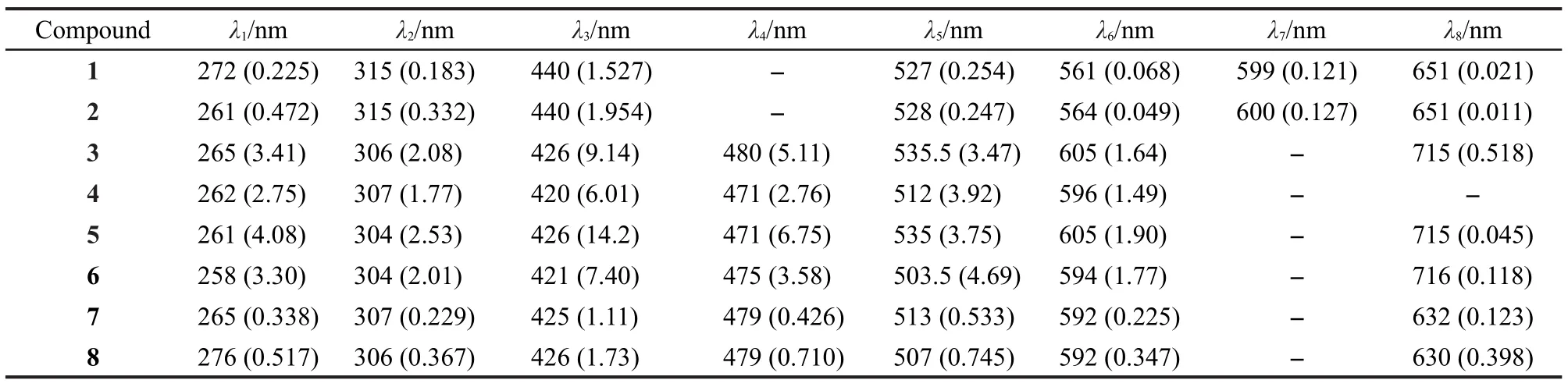
表2 化合物的紫外-可见光谱特征吸收峰数据aTable 2 UV-Vis absorbance data of compoundsa
3.4 荧光光谱
图1为化合物A、2及其金属镍化合物3-6的荧光光谱图.化合物A的激发波长为480 nm,化合物2的激发波长为440 nm,金属镍化合物3-6的激发波长为420 nm.激发狭缝为10 nm,发射狭缝为20 nm.
由荧光发射光谱图可以看出,化合物A和2的氯仿溶液分别在716和724 nm处出现强发射峰,并且化合物2较化合物A的荧光光谱约有8 nm的红移,这是由于3,5-二叔丁基水杨醛的醛基与化合物A中的胺基发生缩合反应后,取代基上的电子参与了整个体系的共轭大π键,增大了共轭体系,使整个卟啉共轭体系上的电子跃迁能级降低所致.43

图1 化合物A、2及3-6在氯仿中的荧光发射光谱图Fig.1 Fluorescence spectra of compoundsA, 2,and 3-6 in chloroform
卟啉的共轭体系中连有供电子取代基会使卟啉的荧光强度增加,反之,连有吸电子取代基则会使荧光强度明显降低.供电子基中含有未成键电子,而未成键电子可以激发转移到卟啉环上.由于未成键电子的电子云为不等性的sp3杂化,几乎与卟啉环上的π轨道平行,因而未成键电子可以参与卟啉环π电子的共轭,即扩大其共轭体系.这样就使得供电子基取代的卟啉化合物的吸收光谱和发射光谱波长比未取代的原卟啉化合物要长,荧光效率也有提高.当卟啉环外侧连有吸电子基时,虽然吸电子基中也含有未成键电子,但是未成键电子并不参与卟啉环共轭π键.因为吸电子基中的未成键电子跃迁到π*键上时,其轨道与π*轨道的空间分布差异很大,几乎不交叠,未成键电子要从空间的一个方向(即其本身的轨道)向另一方向(π*轨道)跃迁,这是一种“禁阻跃迁”(跃迁的几率很小),致使其荧光减弱,或者说对荧光具有一定的抑制作用.44化合物A的结构中含有两个具有供电子效应的―NH2,两个―NH2上的未成键电子的电子云与卟啉环的π轨道形成共轭,使得化合物A比化合物2的荧光要强.
当金属Ni2+离子与卟啉配体之间发生配位以后,测定其荧光发射光谱,均出现荧光淬灭现象.这是由于金属镍属于过渡金属,Ni3+、Ni+离子具有顺磁性,顺磁性金属离子的第一激发单重态(S1)同第一激发三重态(T1)之间存在系间窜跃的能量衰减形式.由于顺磁性粒子的存在,加强了体系中带电分子、极性分子和可极化分子的不规则运动所产生的瞬间磁效应,这种磁效应的微扰可与电子自旋耦合,从而改变自旋方向,使系间窜跃显著增大.同时这种微扰作用也会对分子结构造成影响,使得激发分子与溶剂分子相互作用增加,造成能量损失的增加.另外,由于镍离子的激发态能级(m*)介于其S1和T1之间,发生了S1→m*→T1及S1→T1的非辐射跃迁,而此过程也造成了能量的损失.正是以上的多种原因造成了卟啉金属镍配合物荧光的淬灭.45
4 结论
采用1H NMR、红外光谱、紫外-可见光谱、荧光及质谱等方法对Salen-卟啉型配体及单、双核金属配合物的研究表明,本文合成的目标化合物所包含的卟啉和Salen部分是共轭相连,整个分子具有π共轭骨架结构.金属离子与分子中的两个配位空腔的分别配位或双配位,形成了单核镍、双核镍和异双核镍、锌金属配合物.单核镍及异双核镍、锌配合物中,镍离子落入Salen部分的配位空腔,而锌离子则是与卟啉部分形成锌卟啉大环结构.Salen-卟啉型配体的荧光由于金属离子的配位而出现猝灭现象.
(1)Andruh,M.Chem.Commun.2007,No.25,2565.
(2) McInnes,E.J.L.;Piligkos,S.;Timco,G.A.;Winpenny,R.E.P. Coord.Chem.Rev.2005,249(23),2577.doi:10.1016/j.ccr. 2005.02.003
(3) Cooke,M.W.;Hanan,G.S.Chem.Soc.Rev.2007,36(9),1466. doi:10.1039/b609200b
(4) Beltran,L.M.C.;Long,J.R.Accounts Chem.Res.2005,38(4), 325.doi:10.1021/ar040158e
(5) Manoli,M.;Prescimone,A.;Bagai,R.;Mishra,A.;Murugesu, M.;Parsons,S.;Wernsdorfer,W.;Christou,G.;Brechin,E.K. Inorg.Chem.2007,46(17),6968.doi:10.1021/ic7007528
(6) Beletskaya,I.;Tyurin,V.S.;Tsivadze,A.Y.;Guilard,R.;Stern, C.Chem.Rev.2009,109(5),1659.doi:10.1021/cr800247a
(7) Jin,B.;Lee,H.M.;Lee,Y.A.;Ko,J.H.;Kim,C.;Kim,S.K. J.Am.Chem.Soc.2005,127(8),2417.doi:10.1021/ja044555w
(8) Balaban,T.S.Accounts Chem.Res.2005,38(4),612.
(9)Elemans,J.A.A.W.;Hameren,R.;Nolte,R.J.M.;Rowan,A. E.Adv.Mater.2006,18(10),1251.doi:10.1002/adma. 200502498
(10) Imahori,H.J.Phys.Chem.B 2004,108(20),6130.doi: 10.1021/jp038036b
(11)Zhang,X.H.;Jiao,Z.;Yan,W.W.;Ruan,W.J.;Zhu,Z.A.Acta Phys.-Chim.Sin.2010,26,701. [张晓红,矫 志,闫伟伟,阮文娟,朱志昂.物理化学学报,2010,26,701.]doi:10.3866/ PKU.WHXB20100306
(12)Ren,X.F.;Ren,A.M.;Wang,Q.;Feng,J.K.Acta Phys.-Chim. Sin.2010,26,110.[任雪峰,任爱民,王 钦,封继康.物理化学学报,2010,26,110.]doi:10.3866/PKU.WHXB20100103
(13) Song,C.E.;Lee,S.G.Chem.Rev.2002,102(10),3495.doi: 10.1021/cr0103625
(14) Katsuki,T.Synlett 2003,No.3,281.
(15)Venkataramanan,N.S.;Kuppuraj,G.;Rajagopal,S.Coord. Chem.Rev.2005,249(11-12),1249.doi:10.1016/j.ccr. 2005.01.023
(16) Jin,C.;Jia,Y.J.;Wang,B.J.;Fan,B.B.;Ma,J.H.;Li,R.F. Acta Phys.-Chim.Sin.2006,22,947.[晋 春,贾银娟,王宝俊,范彬彬,马静红,李瑞丰.物理化学学报,2006,22,947.] doi:10.3866/PKU.WHXB20060808
(17) Lu,Z.;Yuan,M.;Pan,F.;Gao,S.;Zhang,D.;Zhu,D.Inorg. Chem.2006,45(9),3538.doi:10.1021/ic051648l
(18) Lacroix,P.G.Eur.J.Inorg.Chem.2001,No.2,339.
(19) Binnemans,K.Chem.Rev.2005,105(11),4148.doi:10.1021/ cr0400919
(20) Liu,J.;Guo,L.Q.;Zhang,X.H.;Ruan,W.J.;Zhu,Z.A.Acta Phys.-Chim.Sin.2012,28,265. [刘 佳,郭莉芹,张晓红,阮文娟,朱志昂.物理化学学报,2012,28,265.]doi:10.3866/ PKU.WHXB201111251
(21) Guo,L.Q.;Shi,X.L.;Ruan,W.J.;Zhang,X.H.;Zhu,Z.A. Acta Phys.-Chim.Sin.2010,26,1195.[郭莉芹,史秀丽,阮文娟,张晓红,朱志昂.物理化学学报,2010,26,1195.]doi: 10.3866/PKU.WHXB20100336
(22) Leadbeater,N.E.;Marco,M.Chem.Rev.2002,102(10),3217. doi:10.1021/cr010361c
(23) Drain,C.M.;Varotto,A.;Radivojevic,I.Chem.Rev.2009,109 (5),1630.doi:10.1021/cr8002483
(24) Perrin,D.D.;Armarego,W.L.F.;Perrin,D.R.Purification of Laboratory Chemicals,2nd ed.;Chemical Laboratory Press: Beijing,1987;p 126;translated by Shi,Y. [Perrin,D.D.; Armarego,W.L.F.;Perrin,D.R.实验室化学药品的提纯方法.第二版.时 雨,译.北京:化学工业出版社,1987:126.]
(25) Kadish,K.M.;E,W.;Sintic,P.J.;Ou,Z.;Shao,J.;Ohkubo,K.; Fukuzumi,S.;Govenlock,L.J.;McDonald,J.A.;Try,A.C.; Cai,Z.L.;Reimers,J.R.;Crossley,M.J.J.Phys.Chem.B 2007,111(30),8762.doi:10.1021/jp0726743
(26)Thordarson,P.;Marquis,A.;Crossley,M.J.Org.Biomol.Chem. 2003,1(7),1216.doi:10.1039/b211015f
(27) Reek,J.N.;Rowan,A.E.;Crossley,M.J.;Nolte,R.J.J.Org. Chem.1999,64(18),6653.doi:10.1021/jo990348g
(28)Wang,C.Z.Study on Molecular Recognition,Asymmetric Catalysis and Theoretic Chemistry of Metalloporphyrins.Ph.D. Dissertation,Nankai University,Tianjin,2000.[王传忠.金属卟啉的分子识别、不对称催化及理论研究[D].天津:南开大学,2000.]
(29)Wang,N.X.Nuclear Magnetic Resonance Spectroscopy; Chemical Industry Press:Beijing,2006;pp 15-25. [王乃兴.核磁共振谱学.北京:化学化工出版社,2006:15-25.]
(30)Kruper,W.J.;Chamberlin,T.A.;Kochanny,M.J.Org.Chem. 1989,54(11),2753.doi:10.1021/jo00272a057
(31) Gouterman,M.J.Chem.Phys.1959,30(5),1139.doi:10.1063/ 1.1730148
(32)Adler,A.D.;Longo,F.R.;Finarelli,J.D.;Goldmacher,J.; Assour,J.;Korsakoff,L.J.Org.Chem.1967,32(2),476.
(33)Yeow,E.K.L.;Steer,R.P.Phys.Chem.Chem.Phys.2003,5 (1),97.
(34) Wang,D.;Zhang,J.;Shi,T.;Wang,B.;Cao,X.;Li,T. J.Photochem.Photobiol.A 1996,93(1),21.doi:10.1016/ 1010-6030(95)04142-7
(35) Gouterman,M.In The Porphyrins;Dolphin,D.Ed.;Academic Press:New York,1979;Vol.III,PartA,pp 1-156.
(36) Sacconi,L.;Ciampolini,M.;Maggio,F.;Cavasino,F.P.J.Am. Chem.Soc.1962,84(17),3246.doi:10.1021/ja00876a005
(37) Carlin,R.L.Transition Metal Chemistry;Marcel Dekker,New York,1965,p 239.
(38) Tas,E.;Aslanoglu,M.;Kilic,A.;Kara,Z.J.Coord.Chem. 2006,59(8),861.doi:10.1080/00958970500412206
(39) Odabaşoğlu,M.;Arslan,F.;Ölmez,H.;Büyükgüngör,O.Dyes Pigm.2007,75(3),507.doi:10.1016/j.dyepig.2006.06.033
(40) Chen,Z.;Wu,Y.;Gu,D.;Gan,F.Spectrochim.Acta Part A 2007,68(3),918.doi:10.1016/j.saa.2007.01.006
(41)Chen,Z.;Huang,F.;Wu,Y.;Gu,D.;Gan,F.Inorg.Chem. Commun.2006,9(1),21.doi:10.1016/j.inoche.2005.10.006
(42) Kasumov,V.T.Spectrochim.Acta Part A 2003,57(8),1649.
(43)Zhang,H.S.;Wang,H.;Zhao,Y.Y.Molecular Probes and Detection Reagents;Science Press:Beijing,2002;pp 25-98.[张华山,王 红,赵媛媛.分子探针与检测试剂.北京:科学出版社,2002:25-98.]
(44) Hariman,A.;Odabaşoğlu,M.;Arslan,F.;Ölmez,H.; Büyükgüngör,O.J.Chem.Soc.Faraday Trans.1 1981,77(2), 369.doi:10.1039/f19817700369
(45) Penzer,G.R.Molecular Luminescence Analytical Methods (Fluorescence and Phosphorescence Methods);Fudan University Press:Shanghai,1985;p 10;translated by Zhu,D. C.,Chen,J.H.,Zhu,S.S.[Penzer,G.R.分子发光分析法(荧光法和磷光法).祝大昌,陈剑宏,朱世盛,译.上海:复旦大学出版社,1985:10.]
猜你喜欢
中学生数理化(高中版.高考数学)(2022年1期)2022-04-26
化学工程师(2022年3期)2022-04-19
房地产导刊(2022年1期)2022-02-28
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
上海化工(2021年2期)2021-04-23
当代陕西(2019年6期)2019-04-17
计算机测量与控制(2017年6期)2017-07-01
科技与企业(2015年20期)2015-10-21
外语学刊(2014年3期)2014-12-03
郑州大学学报(理学版)(2013年2期)2013-03-11
