
Theoretical lnvestigation of Structure-Sensitivity of Styrene Epoxidation on Ag(111)and Ag(110)Surfaces
2013-07-25 09:11WANGChenWEIZiZhangLYongKangXINGBinWANGGuiChang
物理化学学报 2013年4期
WANG Chen WEI Zi-Zhang LYong-Kang,* XING Bin WANG Gui-Chang,*
(1Key Laboratory of Coal Science and Technology,Ministry of Education and Shanxi Province,Taiyuan University of Technology,Taiyuan 030024,P.R.China; 2Tianjin Enviromental Engineering Assessment Center,Tianjin 300191,P.R.China;3College of Chemistry and Tianjin Key Laboratory of Metal and Molecule-based Material Chemistry,Nankai University,Tianjin 300071,P.R.China)
1 lntroduction
The selective epoxidation of terminal alkene on Ag catalyst has received a number of experimental and theoretical studies because of its technological importance.1-9Linicet al.5,6,10have examined the elementary steps of the selective ethylene oxidation,in which they reported that the oxametallacycle intermediate(OMME)plays a key role in controlling the reaction activity and selectivity.The experimental and theoretical studies indicated that the epoxdiation process of ethylene on Ag(100)is more favorable than that on Ag(111)surface,11and the possible reason why the energy barrier for the ethylene epoxide(EO)formation on Ag(100)surface is smaller than that on Ag(111)surface comes from the fact that the Ag(100)binds CH3O more strongly,and thus results in the more energy required to elongate the Ag-O bond to form acetaldehyde(AC).Similarly,Chenet al.12studied the ethylene epoxidation process on Au catalyst and reported that the process on Au nanoparticles are more selective than that on Au(111)surface,i.e.,a structure sensitivity reaction.The reason behind such behave is the fact that a late transition state(TS)(i.e.,product-like)for the epoxidation process is found on nanoparticle or(100)surface,and an early TS(i.e.,reactant-like)is found on the flat(111)surface.These studies demonstrate that the stronger adsorption of oxametallacycle on surface,the larger energy barrier difference between EO and AC formation will be,and thus the higher selectivity towards the EO formation.Barteau and Madix13studied ethylene epoxidation under ultra-high vacuum(UHV)condition,and they reported a quite low reactivity for ethylene and oxygen atoms on Ag(110)surface.This is related to the weak adsorption of ethylene,which means that the ethylene molecule may desorb from the surface before the reaction happens.To solve this limitation,higher molecular weight terminal alkene with higher desorption barrier can be used.For instance,norbornene,14styrene,15-20and 3,3-dimethyl-1-butene21have been used to study the epoxidation process of olefins under UHV conditions.Styrene selective oxidation on IB group metals22has also been studied in the past few years due to its similar properties like ethylene,for example,it also does not contain allylic C-H bond.23Moreover,the interaction between molecular styrene with substrate may be stronger than that of molecular ethylene due to the existence of phenyl ring in styrene,which would lead to the long lifetime of styrene on surface and thus a higher catalytic activity can be expected.Quilleret al.23studied the selective oxidation of styrene on oxygen-covered Au(111)surface under the UHV condition.In their study,they found that the final produce distribution was significantly affected by the reaction coverage,and the highest selectivity for styrene oxide formation occurred at the situation where the styrene coverage was 0.28 ML and oxygen coverage was 0.20 ML.In particular,recently Zhou and Madix8demonstrated that the styrene epoxidation on Ag single crystal was a highly structure-sensitive reaction,and the linear oxametallacycle(i.e.,the pre-adsorbed atomic oxygen bound to the methylene group in styrene)was the major intermediate.They found that the selectivity to the epoxidation process was higher on(111)surface than that on(110)surface,2,18-20and a possible reaction mechanism for styrene epoxidation on Ag(110)and Ag(111)surfaces was proposed8(see Scheme 1).On Ag(110)surface,most of the intermediates are transformed to combustion intermediate and form different oxidation products,which are phenylacetaldehyde and phenylketene.8,19In contrast,the major product is styrene oxide on Ag(111)surface.2,8,18,20Although possible reaction mechanism is proposed by the experimental study to account for the structure-sensitivity,the further confirmation by theoretical study is required.In the present work,in order to provide further understanding of the structure sensitivity of styrene epoxidation on Ag catalyst,the density functional theory(DFT)calculations have been performed,and then the chemical kinetics simulation was utilized to further compare with the experimental findings.
2 Calculation methods and models
To study the energy and structure details of styrene epoxidation,the periodic,self-consistent DFT calculation was performed by using the ViennaAb-initioSimulation Package(VASP).24,25The electronic structures were calculated using DFT within GGA-PW91 functional.26The projector-augmented wave(PAW)27,28scheme was used to describe the interaction between the valence electrons and the inner cores,and the electronic spatial wave-function was expended in a plane wave basis with kinetic cut-off energy of 400 eV.The nudged elastic band(NEB)was employed to locate the transition state(TS).29When an approximate saddle point was obtained by the nudged elastic band calculation,the quasi-Newton algorithm was then used to refine it by minimizing residual forces for all unconstrained atoms below 0.5 eV·nm-1.At last,the frequency analysis was performed to confirm the TS by verifying the existence of the only one imaginary frequency.The surfaces were modeled by a periodic slab ofp(4×4)supercell containing three layers of atoms with a relaxation of the uppermost layer.The Monkhorst-Pack mesh of 3×3×1 specialk-point was used to sample in the Brillouin zone.30The optimized lattice constant of 0.418 nm forAg was used.
3 Results and discussion
3.1 High oxygen coverage model

Scheme 1 Competitive reaction for styrene oxidation and corresponding reaction barriers(eV)onAg(110)andAg(111)surfaces
Here the physisorption and dissociation of the oxygen molecule on the surfaces are not explicitly considered,so the process is studied with the surfaces with pre-adsorbed oxygen.On the Ag(110)surface,the pre-adsorbed oxygen atom prefers the 3-fold hollow site(3h)with the adsorption energy of-3.59 eV.On Ag(111)surface,the most stable adsorption site for pre-adsorbed oxygen atom is the fcc site,and the adsorption energy is-3.69 eV.To represent high oxygen coverage of 0.375 monolayer(ML)8observed in the experimental condition,six oxygen atoms were placed on both Ag(110)and Ag(111)surfaces.Several possible configurations to present the high oxygen coverage were examined,and it was found that the most stable configuration is the case where each oxygen atom adsorbed at its most stable adsorption site(see Fig.S1 in the Supporting Information).Since more oxygen atoms present on the Ag surface,one may expect that the adsorption energy of the oxygen atom may change due to the lateral interaction between oxygen atoms.The average adsorption energy for each oxygen atom can be calculated as:31
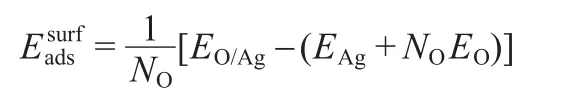
whereNOis the number of oxygen atoms,EO/Ag,EAg,andEOare the total energies of adsorption system,the clean surface,and the free atomic oxygen,respectively.The average oxygen atom adsorption energies on Ag(110)and Ag(111)surfaces are calculated to be-3.47 and-3.31 eV,respectively,which are smaller than that on the low oxygen coverage as discussed above.In order to confirm the most stable existing form of oxygen atoms,the pre-adsorbed atomic oxygen on Ag(111)surface and the lattice oxygen in surface oxide structure have been compared,32and the adsorption energy for lattice oxygen can be calculated as:

whereNAgis the number of missing Ag atom in the oxide structure compared to ideal Ag(111)surface and,repersents the energy of Ag atom in bulk.In this paper,NOis 6 and the corresponding oxide structure is Ag11O633(see Fig.S1,Supporting Information),so theNAgis 5.The calculated adsorption energy of oxygen in oxide structure is-1.94 eV,which is lower than the average adsorption energy of atomic oxygen.This indicates that the surface chemisorbed oxygen atom on Ag(111)surface is the energetically favorable configuration rather than the surface oxide structure.So the oxygen atom pre-covered Ag(111)model instead of the surface metal oxide model was used in this work.It should be pointed here that our present calculation result is different from Shi and Stampfl's finding32on metallic Au,in which they reported that the thin surface-oxide-like structure with an oxygen coverage of 0.3125 ML(Au6O5)is more stable.The possible reason may be due to the different activity between Ag and Au,that is,oxygen prefers to chemisorb on more active metal surface(Ag)and forms surface oxide on less active metal surface(Au).
3.2 Adsorption properties of possile species
From Scheme 1 we know that there are five different products based on the reaction path I and path II,namely styrene oxide(C8H8O),combustion intermediate(C6H5CHCHO),benzaldehyde(C6H5CHO),phenyl acetaldehyde(C6H5CH2CH2O),and phenylketene(C6H5CHCO).Table 1 gives the calculated adsorption properties of possible species,and the corresponding adsorption configurations are displayed in Fig.S2(Supporting Information).
3.2.1 Styrene

Table 1 Adsorption energies and geometry parameters for reactant,oxametallacycle intermediate,and products on oxygen pre-adsorbed Ag(110)andAg(111)surfaces
The adsorption energies of styrene are found to be-0.09 and-0.04 eV on Ag(110)and Ag(111)surfaces with the oxygen coverage of 0.375 ML,respectively.In the optimized configuration,the styrene molecule is nearly parallel to the surface.The styrene molecule is about 0.350 and 0.377 nm away from theAg(110)andAg(111)surfaces,respectively.
3.2.2 Oxametallacycle(OMMS)
Due to the asymmetry of the styrene molecule,there are two possible reaction intermediates in the styrene epoxidation processes.34,35In this paper,the carbon atom is numbered as C6H5-C1H=C2H2.Correspondingly,the branched oxametallacycle configuration is Ag-O-C1-C2-Ag and the Ag-C1-C2-O-Ag will be designated as linear oxametallacycle.After optimization,the two oxametallacycles on Ag(110)surface and branched oxametallacycle on Ag(111)surface are the closedring structure,which is consistent with the case of ethylene epoxidation on Cu(111)surface.36,37But the linear oxametallacycle on Ag(111)surface is an open-ring type,which is similar to the structure on Au(111)surface.35The shortest distance between C1and surface Ag atom is 0.336 nm and the O…Ag distance is 0.231 nm.
3.2.3 Styrene oxide(C8H8O)
The adsorption energies of styrene epoxide on Ag(110)and Ag(111)surfaces are found to be-0.05 and-0.22 eV,respectively.And the phenyl group is nearly parallel to the Ag surface,which is similar to that of the styrene molecule.The plane of the C1-C2-O ring structure is about perpendicular to the surface.
3.2.4 Benzaldehyde(C6H5CHO)
The benzaldehyde is formed by methylene dissociation of branched oxametallacycle,and the optimized adsorption structure is very similar with the free benzaldehyde molecule.The adsorption energy is-0.01 eV on Ag(110)surface and-0.39 eV onAg(111)surface.
3.2.5 Phenyl acetaldehyde(C6H5CH2CHO)
The phenyl acetaldehyde is the by-product of the favorable reaction pathvialinear oxametallacycle,which is the result of H transfer from C2to C1.The adsorption structure of phenyl acetaldehyde on Ag(110)and Ag(111)surfaces are almost the same,which are both similar with free phenyl acetaldehyde molecule and the distance between the molecule and the surface are also close.The adsorption energy is-0.11 eV on Ag(110)surface and-0.27 eV onAg(111)surface.
3.2.6 Combustion intermediate(C6H5CHCHO)
Previously,Zhou and Madix8reported the existence of combustion intermediate during styrene epoxidation on Ag single crystal.The plane of benzene ring of the optimized combustion intermediate structure is a little tilted toward the surface with the dihedral angle being about 20°on Ag(110)surface,while it is only about 15°onAg(111)surface.
3.2.7 Phenylketene(C6H5CHCO)
The phenylketene is formed by hydrogen atom elimination of combustion intermediate,and the phenylketene molecule is basically in the same plane with C1- C2=O in a line.On Ag(110)surface,the C1- C2=O line is a little tilted down,which is opposite to that on Ag(111)surface(see Fig.S2).The adsorption energy is-0.18 eV on Ag(110)surface and-0.43 eV onAg(111)surface.
In order to analyze the adsorption properties of different species on the surface,the electronic property analysis is performed.Fig.S3 shows the projected density of states(PDOS)onto adsorbed complex molecular orbital for branched oxametallacycle,linear oxametallacycle,styrene oxide,and phenyl acetaldehyde on Ag(110)surface.One can find that a good liner correlation exists between the highest occupied molecular orbital(HOMO)energy level and the adsorption energy(Fig.1).That is,the lower energy of the HOMO,the stronger the adsorption will be.The reason why the phenyl acetaldehyde has the relatively low HOMO level compared to other species such as linear oxametallacycle is possible due to the more electron transfer between phenyl acetaldehyde and Ag(110)as its much strong interaction with theAg(110)(Table 1).

Fig.1 Relationship between the HOMO energy level and the adsorption energies of different species onAg(110)surface
3.3 Epoxidation of styrene on Ag(110)surface
The calculated reaction energies for epoxidation of styrene on Ag(110)surface are given in Table 2.The detailed of reaction coordinate and corresponding TS configuration are shown in Fig.S4 and Fig.S5b.For the reaction path I,the branched oxametallacycle formation is the rate-controlling step with the energy barrier of 1.31 eV,which is much larger than of the cyclization process of branched oxametallacycle to form styrene oxide(0.16 eV).The energy barrier for the side reaction is 1.54 eV,which means that the selectivity to the formation of benzaldehyde is quite low.The formation of linear oxametallacycle during the reaction path II is favored than the formation of branched oxametallacycle both kinetically(0.21 eVvs1.31 eV)and thermodynamically(-0.78 eVvs0.50 eV).There are three different reactions that start from linear oxametallacycle,that is,the ring-closure to form styrene oxide(0.43 eV),the C2-H elimination to produce combustion intermediate(0.40 eV),and the C2-H transfer to C1to form phenyl acetaldehyde(0.31 eV).The combustion intermediate can further produce the phenylketene by eliminating the second C2-H(0.93 eV).The calculated energy barrier is consistent with the experimental results to a certain extent,that the main products are phenyl acetaldehyde and phenylketene instead of styrene oxide.8
3.4 Epoxidation of styrene on Ag(111)surface
The calculated reaction energies for epoxidation of styrene on Ag(111)surface are given in Table 2.The detailed of reaction coordinate and corresponding TS configuration are shown in Fig.S4 and Fig.S5a.The reaction path I is similar to that on the Ag(110)surface,while the formation of branched oxametallacycle is still the rate-controlling step compared to the formation of styrene oxide(0.42 eVvs0.19 eV).Also the formation of benzaldehyde is difficult due to the much higher energy barrier of 1.34 eV.For the reaction path II,the formation of linear oxametallacycle becomes the rate-controlling step(0.34 eV),while the energy barrier for the following reaction step is lower(0.02,0.01,and 0.21 eV corresponding to different products of styrene,combustion intermediate,and phenyl acetaldehyde).Though the energy barrier for the formation of combustion in-termediate is very small,the following reaction is difficult to perform as a result of higher energy barrier(1.02 eV).

Table 2 Reaction mechanism of styrene epoxidation and the related TS properties
Lukaskiet al.38studied the reaction step of linear oxametallacycle→styrene epoxide by temperature-programmed reaction and DFT calculations.They found that the reaction barrier for the formation of styrene epoxide is 1.43 eV on Ag13cluster and 1.34 eV on Ag10cluster,and it is an endothermic reaction.This is much higher than our calculated results.The possible reason may be the different models and calculation methods used and the contribution of dispersion effect.Because the DFT-GGA calculations may underestimate the adsorption energy of weakly adsorbed molecule due to the lack of long-range interaction(i.e.,dispersion effect).Indeed,the general correction of dispersion effect to the adsorption energy of aromatic molecules on noble metals is ca 0.60 eV39based on the scheme of Grimmeet al.40,41If the dispersion correction was taken into account in the estimation of oxametallacycle adsorption energy,then the activation energy will be increased by ca 0.60 eV,and thus difference between our results and that of Lukaskiet al.will become small.It should be pointed that the effect of dispersion force on the activation energy is very complex because the dispersion force not only affects the reactant but also affects the transition state,and the exactly activation energy calculation should be taken by considering the dispersion force for both the reactant and transition state.42
3.5 Comparison between Ag(110)and Ag(111)surfaces
On both Ag(110)and Ag(111)surfaces,the styrene epoxidation processvialinear oxametallacycle is found to be the favorable pathway,which is consistent with the experimental results that the pre-adsorbed oxygen atom mainly interacts with the-CH2group(i.e.,linear oxametallacycle)in the selective oxidation of styrene on oxygen covered Ag catalyst surface.8,19,20In addition,the present conclusion is very similar to the previous DFT calculation result that the reaction pathvialinear oxametallacycle is more kinetically favored than thatviabranched oxametallacycle for the propylene epoxidation on Cu(111)and Ag(111)surfaces43and styrene epoxidation on Cu(111)and Au(111)surfaces.34,35
It is important to analyze the reaction processvialinear oxametallacycle,which may explain the activity and the selectivity of styrene epoxidation on different Ag surfaces.From comparison of the energetic results of different reaction stepsvialinear oxametallacycle on Ag(111)and Ag(110)surfaces(see Scheme 1),it is general that the Ag(111)surface has lower energy barrier compared to the Ag(110)surface.The possible reason may come from the fact that the average adsorption energy of oxygen atom on Ag(110)surface is higher than that on Ag(111)surface(i.e.,-3.47 eV on Ag(110)surface and-3.31 eV on Ag(111)surface),which means that reactants are hard to be activated by atomic oxygen on Ag(110)surface.Moreover,for the formation of different productsvialinear oxametallacycle(styrene epoxide,combustion intermediate,and phenyl acetaldehyde),the reaction barriers are obviously different.On Ag(110)surface,they are 0.43,0.40,and 0.31 eV,while on Ag(111)surface,they are 0.02,0.01,and 0.21 eV,respectively.It can be concluded that the selectivity towards styrene epoxide on Ag(111)surface should be higher than that on Ag(110)surface qualitatively.In conclusion,the styrene epoxidation on silver is a structure sensitive reaction,and Ag(111)surface is more beneficial for the formation of styrene epoxide than Ag(110)surface.It is necessary to compare present results with the case of ethylene epoxidation process.For the process of ethylene epoxidation,the Ag(100)surface shows higher selectivity than the Ag(111)surface due to the energy barrier difference between AC and EO on(100)surface larger than(111)surface,11whereas an reverse trend found for styrene epoxidation(see Table 2).The possible reason may be due to the different properties of TSs:“late TS”for the ethylene epoxidation process is found on(100)surface and an“early TS”is found on the flat(111)surface,but an“early TS”is found on both Ag(111)andAg(110)surfaces for styrene epoxidation(see Supporting Information),which may lead to the different kinetic behavious for ethylene and styrene epoxidation.
The analysis of the contribution of the atomic Agd-states during the reaction process can explain the reaction selectivity in the production of styrene oxide,phenyl acetaldehyde,and combustion intermediate.The stabilization energy(Es)of thed-state due to its interactions with other orbitals in the TS complex can be calculated by the formula:
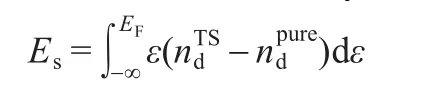

Fig.2 Micro-kinetic simulation of tryrne epoxidation onAg(111)andAg(110)surfaces
At last,to make a quantitative analysis and compare with the experimental findings more directly,the micro-kinetic analysis of the styrene epoxidation has been also performed(see Supporting Information for detail).Fig.2 shows the temperaturedependence of relative selectivity for epoxidation process on Ag(111)and Ag(110)surfaces.The relative selectivity of styrene epoxidation on Ag(111)surface(0.38)is much higher than that on Ag(110)surface(0.003),and the reaction rate of styrene oxide formation on Ag(111)surface is 10-5site-1·s-1at the reaction temperature of 280 K.The possible reason for the lower selectivity on Ag(110)surface is that the energy barrier for the styrene epoxidation formation is higher than the formation of phenyl acetaldehyde and combustion intermediate(Scheme 1 as well as Table 2).
4 Conclusions
In conclusion,the present DFT study has clearly reproduced the experimental observation for the selective oxidation of styrene on Ag(110)and Ag(111)surfaces.The formation of oxametallacycle intermediate is an important reaction step in the styrene epoxidation process.The calculated results show that the formation of styrene oxideviathe linear oxametallacycle is kinetically more favorable than that of branched oxametallacycle on both Ag(110)and Ag(111)surfaces.The DFT results indicate that the combustion intermediate is more stable on Ag(110)surface than on Ag(111)surface.The micro-kinetic simulation results indicate a higher selectivity towards the epoxidation process onAg(111)surface.
(1) Barteau,M.A.Surf.Sci.2006,600,5021.doi:10.1016/j.susc.2006.09.024
(2) Klust,A.;Madix,R.J.Surf.Sci.2006,600,5025.doi:10.1016/j.susc.2006.08.049
(3) Serafin,J.G.;Liu,A.C.;Seyedmonir,S.R.J.Mol.Catal.A:Chem.1998,131,157.doi:10.1016/S1381-1169(97)00263-X
(4) Bocquet,M.L.;Sautet,P.;Cerda,J.;Carlisle,C.I.;Webb,M.J.;King,D.A.J.Am.Chem.Soc.2003,125,3119.doi:10.1021/ja027634l
(5) Linic,S.;Barteau,M.A.J.Am.Chem.Soc.2002,124,310.doi:10.1021/ja0118136
(6) Linic,S.;Barteau,M.A.J.Am.Chem.Soc.2003,125,4034.doi:10.1021/ja029076g
(7) Park,D.M.;Ghazali,S.;Gau,G.Appl.Catal.1983,6(2),175.doi:10.1016/0166-9834(83)80263-2
(8) Zhou,L.;Madix,R.J.Surf.Sci.2009,603,1751.doi:10.1016/j.susc.2008.08.032
(9) Bocquet,M.L.;Loffreda,D.J.Am.Chem.Soc.2005,127,17207.doi:10.1021/ja051397f
(10) Linic,S.;Piao,H.;Adib,K.;Barteau,M.A.Angew.Chem.Int.Edit.2004,43,2918.
(11) Christopher,P.;Linic,S.J.Am.Chem.Soc.2008,130,11264.doi:10.1021/ja803818k
(12) Chen,H.T.;Chang,J.G.;Ju,S.P.;Chen,H.L.J.Phys.Chem.Lett.2010,1,739.doi:10.1021/jz900469f
(13) Barteau,M.A.;Madix,R.J.Surf.Sci.1981,103(2-3),L171.
(14) Roberts,J.T.;Madix,R.J.J.Am.Chem.Soc.1988,110,8540.doi:10.1021/ja00233a038
(15) Lambert,R.M.;Williams,F.J.;Cropley,R.L.;Palermo,A.J.Mol.Catal.A:Chem.2005,228(1-2),27.doi:10.1016/j.molcata.2004.09.077
(16)Hawker,S.;Mukoid,C.;Badyal,J.P.S.;Lambert,R.M.Surf.Sci.Lett.1989,219(3),L615.
(17) Williams,F.J.;Bird,D.P.C.;Palermo,A.;Santra,A.K.;Lambert,R.M.J.Am.Chem.Soc.2004,126,8509.doi:10.1021/ja039378y
(18) Klust,A.;Madix,R.J.J.Am.Chem.Soc.2006,128,1034.doi:10.1021/ja054845s
(19)Liu,X.Y.;Klust,A.;Madix,R.J.;Friend,C.M.J.Phys.Chem.C2007,111,3675.doi:10.1021/jp066560n
(20) Zhou,L.;Madix,R.J.J.Phys.Chem.C2008,112,4725.doi:10.1021/jp7119558
(21) Mukoid,C.;Hawker,S.;Badyal,J.P.S.;Lambert,R.M.Catal.Lett.1990,4(1),57.doi:10.1007/BF00764871
(22)Deng,X.Y.;Friend,C.M.J.Am.Chem.Soc.2005,127,17178.doi:10.1021/ja0557031
(23) Quiller,R.G.;Liu,X.Y.;Friend,C.M.Chemistry-An Asian Journal2010,5(1),78.doi:10.1002/asia.v5:1
(24) Kresse,G.;Furthmüller,J.Comp.Mater.Sci.1996,6,15.doi:10.1016/0927-0256(96)00008-0
(25) Kresse,G.;Hafner,J.Phys.Rev.B1994,49,14251.doi:10.1103/PhysRevB.49.14251
(26) Perdew,J.P.;Chevary,J.A.;Vosko,S.H.;Jackson,K.A.;Pederson,M.R.;Singh,D.J.;Fiolhais,C.Phys.Rev.B1992,46,6671.doi:10.1103/PhysRevB.46.6671
(27) Kresse,G.;Joubert,D.Phys.Rev.B1999,59,1758.doi:10.1103/PhysRevB.59.1758
(28) Blouml,P.E.Phys.Rev.B1994,50,17953.doi:10.1103/PhysRevB.50.17953
(29) Henkelman,G.;Uberuaga,B.P.;Jónsson,H.J.Chem.Phys.2000,113,9901.doi:10.1063/1.1329672
(30) Monkhorst,H.J.;Pack,J.D.Phys.Rev.B1976,13,5188.doi:10.1103/PhysRevB.13.5188
(31) Li,W.X.;Stampfl,C.;Scheffler,M.Phys.Rev.B2002,65,075407.doi:10.1103/PhysRevB.65.075407
(32) Shi,H.Q.;Stampfl,C.Phys.Rev.B2007,76,075327.doi:10.1103/PhysRevB.76.075327
(33) Bocquet,M.L.;Michaelides,A.;Loffreda,D.;Sautet,P.;Alavi,A.;King,D.A.J.Am.Chem.Soc.2003,125,5620.doi:10.1021/ja0297741
(34)Pang,X.Y.;Xing,B.;Xue,L.Q.;Wang,G.C.J.Comput.Chem.2010,31,1618.
(35)Xue,L.Q.;Pang,X.Y.;Wang,G.C.J.Comput.Chem.2009,30,438.doi:10.1002/jcc.v30:3
(36) Torres,D.;Lopez,N.;Illas,F.;Lambert,R.M.J.Am.Chem.Soc.2005,127,10774.doi:10.1021/ja043227t
(37) Torres,D.;Lopez,N.;Illas,F.J.Catal.2006,243(2),404.doi:10.1016/j.jcat.2006.08.011
(38)Lukaski,A.C.;Enever,M.C.N.;Barteau,M.A.Surf.Sci.2007,601,3372.doi:10.1016/j.susc.2007.06.015
(39) Tonigold,K.;Gross,A.J.Chem.Phys.2010,132,224701.doi:10.1063/1.3439691
(40)Grimme,S.J.Comput.Chem.2006,27,1787.
(41) Grimme,S.;Antony,J.;Ehrlich,S.;Krieg,H.J.Chem.Phys.2010,132,154104.doi:10.1063/1.3382344
(42) Benco,L.;Bucko,T.;Hafner,J.J.Catal.2011,277,104.
(43) Torres,D.;Lopez,N.;Illas,F.;Lambert,R.M.Angew.Chem.Int.Edit.2007,46,2055.
(44) Wang,H.F.;Liu,Z.P.J.Am.Chem.Soc.2008,130,10996.
