
Complete mitochondrial genome of the Five-dot Sergeant Parathyma sulpitia (Nymphalidae: Limenitidinae) and its phylogenetic implications
2013-12-25 01:02TIANLiLiSUNXiaoYanCHENMeiGAIYongHuaHAOJiaShengYANGQun
Zoological Research 2013年3期
TIAN Li-Li, SUN Xiao-Yan, CHEN Mei, GAI Yong-Hua, HAO Jia-Sheng,,*, YANG Qun,*
(1. College of Life Sciences, Anhui Normal University, Wuhu 241000, China; 2. LPS, Institute of Geology and Palaeontology, the Chinese Academy of Sciences, Nanjing 210008, China)
Complete mitochondrial genome of the Five-dot SergeantParathyma sulpitia(Nymphalidae: Limenitidinae) and its phylogenetic implications
TIAN Li-Li1, SUN Xiao-Yan2, CHEN Mei1, GAI Yong-Hua2, HAO Jia-Sheng1,2,*, YANG Qun2,*
(1.College of Life Sciences,Anhui Normal University,Wuhu241000,China; 2.LPS, Institute of Geology and Palaeontology,the Chinese Academy of Sciences,Nanjing210008,China)
The complete mitochondrial genome of theParathyma sulpitia(Lepidoptera, Nymphalidae, Limenitidinae) was determined. The entire mitochondrial DNA (mtDNA) molecule was 15 268 bp in size. Its gene content and organization were the same as those of other lepidopteran species, except for the presence of the 121 bp long intergenic spacer betweentrnS1(AGN) andtrnE. The 13 protein-coding genes (PCGs) started with the typical ATN codon, with the exception of thecox1gene that used CGA as its initial codon. In addition, all protein-coding genes terminated at the common stop codon TAA, except thenad4gene which used a single T as its terminating codon. All 22 tRNA genes possessed the typical clover leaf secondary structure except fortrnS1(AGN), which had a simple loop with the absence of the DHU stem. Excluding the A+T-rich region, the mtDNA genome ofP. sulpitiaharbored 11 intergenic spacers, the longest of which was 121 bp long with the highest A+T content (100%), located betweentrnS1(AGN) andtrnE. As in other lepidopteran species, there was an 18-bp poly-T stretch at the 3'-end of the A+T-rich region, and there were a few short microsatellite-like repeat regions without conspicuous macro-repeats in the A+T-rich region. The phylogenetic analyses of the published complete mt genomes from nine Nymphalidae species were conducted using the concatenated sequences of 13 PCGs with maximum likelihood and Bayesian inference methods. The results indicated that Limenitidinae was a sister to the Heliconiinae among the main Nymphalidae lineages in this study, strongly supporting the results of previous molecular data, while contradicting speculations based on morphological characters.
Parathyma sulpitia; Lepidoptera; Nymphalidae; Limenitidinae; Mitochondrial genome
Insect mitochondrial DNA (mtDNA) is a circular DNA molecule 14-20 kb in size with 13 protein-coding genes (PCGs), two ribosomal RNA genes, 22 tRNA genes, and one A+T-rich region which contains the initiation sites for transcription and replication (Boore, 1999; Clayton, 1992; Wolstenholme, 1992). In recent years, owing to its maternal inheritance, lack of recombination and accelerated nucleotide substitution rates compared to those of the nuclear DNA, the mitochondrial genome has been popularly used in studies on phylogenetics, comparative and evolutionary genomics, population genetics, and molecular evolution.
The Nymphalidae is one of the largest groups of butterflies, comprising about 7 200 described species throughout the world. Its systematic and evolutionary process has long been a matter of controversy (Ackery, 1984, 1999; de Jong et al, 1996; Ehrlich, 1958; Harvey, 1991). Until recently, however, only eight complete or nearly complete mt genome sequences have been determined from Nymphalidae among some forty sequences for Lepidoptera. That is, two from Heliconiinae, two from Satyrinae, and one each from Calinaginae, Apaturinae, Danainae, and Libytheinae.
Limenitidinae is a subfamily of Nymphalidae that includes the admirals and its close relatives. This butterfly group has long been the subject of scientific curiosity, serving as the model organism in diverse fields such as genetics, developmental biology, and evolutionary ecology (Fiedler, 2010; Platt & Maudsley, 1994). However, its sub-group classifications and phylogenetic relationships with the other Nymphalidae groups remains unresolved based on morphological and molecular criteria (Freitas & Brown, 2004; Wahlberg et al, 2003, 2005; Wahlberg & Wheat, 2008; Zhang et al, 2008).
Parathyma sulpitiais a representative species of the subfamily Limenitidinae (Lepidoptera: Nymphalidae) and it is widely distributed in Southeastern Asian areas, such as Vietnam, Burma, India, and China. We determined its complete mitochondrial genome sequence and compared this sequence with those of the other eight-nymphalid butterfly species available. Additionally, we performed phylogenetic analyses using maximum likelihood and Bayesian inference methods based on the concatenated 13 protein coding gene (PCG) sequences. The new sequence data and related analyses may provide useful information about the systematics and evolution of Nymphalidae at the genomic level.
1 Materials and Methods
1.1 Specimen collection
Adult butterflies ofP. sulpitiawere collected from the Jiulianshan National Nature Reserve, Jiangxi Province, China. The specimens were preserved immediately in 100% ethanol and then stored at -20 °C before genomic DNA extraction.
1.2 DNA extraction, PCR amplification and sequencing
Whole genomic DNA was extracted from thoracic muscle tissue with the DNeasy Tissue Kit (Qiagen) after the protocol of Hao et al (2005). Some universal PCR primers for short fragment amplifications of thecox1,cobandrrnLgenes were synthesized (Simon et al, 1994). The remaining short and long primers were designed based on the sequence alignment of the available complete lepidopteran mitogenomes using Primer Premier 5.0 software (Singh et al, 1998).
The entire mitogenome ofP. sulpitiawas amplified in six fragments (cox1-cox3,cox3-nad5,nad5-nad4,nad4-cob,cob-rrnL,rrnL-cox1) using long-PCR techniques with TaKaRa LATaq polymerase under the following cycling conditions: initial denaturation for five minutes at 95 °C, followed by 30 cycles of 95 °C for 50 s, 45-50 °C for 50 s, 68 °C for 2 min and 30 s; and a final extension step of 68 °C for 10 min. The PCR products were visualized by electrophoresis on 1.2% agarose gel, then purified using a 3S Spin PCR Product Purification Kit and sequenced directly with an ABI–377 automatic DNA sequencer. For each long PCR product, the full, double-stranded sequence was determined by primer walking. The mitogenome sequence data were deposited into the GenBank database under the accession number JQ347260.
1.3 Sequence analysis and annotation
The tRNA genes and their secondary structure were predicted using tRNAscan-SE software v.1.21 (Lowe & Eddy, 1997) and the putative tRNA genes, which were not found by tRNAscan-SE, were determined by sequence comparison ofP. sulpitiawith other lepidopterans. The PCGs and rRNAs were confirmed by sequence comparison with ClustalX1.8 software and NCBI BLAST search function (Altschul et al, 1990). Nucleotide composition and codon usage were calculated with DAMBE software (Xia & Xie, 2001).
1.4 Phylogenetic analysis
Multiple sequence alignments of the concatenated sequences the 13 PCGs of the nine nymphalid species with available mitogenomes (Tab. 2) were conducted using Clustal X 1.8 software and then proofread manually (Thompson et al,1997). The phylogenetic trees were constructed using maximum likelihood (ML) (Abascal et al, 2007) and Bayesian inference (BI) (Yang & Rannala, 1997) methods with moth speciesManduca sexta(Cameron & Whiting, 2008) (Tab. 2) used as outgroup. The ML analysis for the nucleotide and amino acid sequences were implemented in the PAUP* software (version 4.0b8) (Swofford, 2002) with TBR branch swapping (10 random addition sequences), the best fitting nucleotide substitution model (GTR+I+Γ) was selected using Modeltest version 3.06 (Posa & Krandall, 1998), and the confidence values of the ML tree were evaluated via the bootstrap test with 100 iterations. The Bayesian analyses were performed using MrBayes 3.1.2 (Ronquist & Huelsenbeck, 2003) with the partitioned strategy, the best fitting substitution model was selected as in the ML analysis; the MCMC analyses (with random starting trees) were run with one cold and three heated chains simultaneously for 1 000 000 generations sampled every 100 generations; Bayesian posterior probabilities were calculated from the sample points after the MCMC algorithm started to converge.
2 Results
2.1 Genome organization
The mitogenome ofP. sulpitiawas a circular molecule 15 268 bp long and consisted of 13 PCGs [cytochrome oxidase subunits 1-3 (cox1-3), NADH dehydrogenase subunits 1-6 and 4L (nad1-6andnad4L), cytochrome oxidase b (cob), ATP synthase subunits 6 and 8 genes (atp6andatp8)], two ribosomal RNA genes for small and large subunits (rrnSandrrnL), 22 transfer RNA genes (one for each amino acid and two for leucine and serine) and a non-coding A+T-rich region. The gene orientation and order of theP. sulpitiamitogenome were identical to those of the other available lepidopteran mitogenomes, except for the presence of the 121 bp long intergenic spacer betweentrnS1(AGN) andtrnE(Tab. 1, Fig. 1). As is the case in many insect mitogenomes, the major strand coded for more genes (nine PCGs and 14 tRNAs) and the A+T-rich region, whereas less genes were coded in the minor strand (four PCGs, eight tRNAs and two rRNA genes).
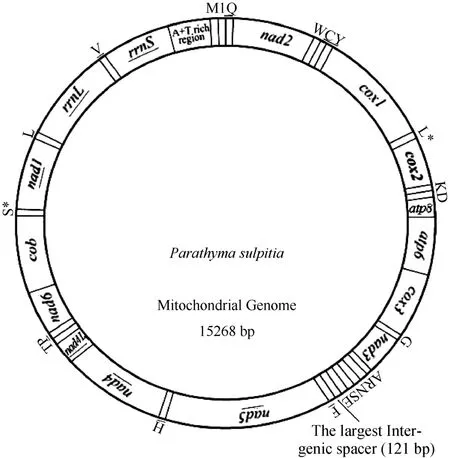
Fig. 1 Circular map of the mitochondrial genome of Parathyma sulpitia
2.2 Protein-coding genes, tRNA and rRNA genes and A+T-rich region
All PCGs in theP. sulpitiamitogenome were initiated by typical ATN codons (seven with ATG, four with ATT, one with ATA), except thecox1gene which was tentatively designated by the CGA codon (Tab. 1). Twelve PCGs ofP. sulpitiahad a common stop codon (TAA), except for thenad4gene which harbored a single T.
The 22 tRNAs varied from 61 [trnCandtrnS1(AGN)] to 71 bp (trnK) in size, and presented typical clover-leaf structure, with the unique exception oftrnS1(AGN), which lacked the dihydrouridine (DHU) stem (Fig. 2). TheP. sulpitiatRNAs harbored a total of 24 pair mismatches in their stems, including six pairs in the DHU stems, eight pairs in the amino acid acceptor stems, two pairs in the TΨC stems and eight pairs in the anticodon stems, respectively. Among these 24 mismatches, 18 were G·U pairs which formed a weak bond in the secondary structure, and the other six were U·U (Fig. 2).

Tab. 1 Summary of the mitogenome of Parathyma sulpitia
As with other insect mitogenome sequences, two rRNA genes (rrnLandrrnS) were detected inP. sulpitia, located betweentrnL1(CUN) andtrnV, and betweentrnVand A+T region, respectively (Fig. 1). The lengths of therrnLand therrnSwere determined as 1 319 bp and 779 bp, respectively.
The A+T-rich region ofP. sulpitiawas 349 bp in size. There was an 18-bp poly-T stretch at the 3'end of the A+T-rich region, and some short microsatellite-like repeat regions without conspicuous macro-repeats throughout the A+T-rich region.
2.3 Phylogenetic analysis
The resultant tree topologies of the ML and Bayesian analyses based on the nucleotide and amino acid sequences were the same, only with a slight difference in their bootstrap support or posterior probability values. For the paper length limit, we have only showed trees based on the nucleotide sequences (Fig. 4) in this paper.

Fig. 2 Predicted secondary clover leaf structures for the 22 tRNA genes of Parathyma sulpitia
3 Discussion
3.1 Genome structure, organization and composition
TheP. sulpitiamitogenome size (15 268 bp) was well within the range detected in the completely sequenced lepidopteran insects, from 15 140 bp inArtogeia melete(GenBank accession no. NC_010568; Hong et al, 2009) to 16 094 bp inAgehana maraho(GenBank accession no. NC_014055; Wu et al, 2010). The nucleotide composition of A+T for theP. sulpitiamitogenome major strand was 81.9%, showing a strongly biased value, which was the highest of all the nymphalid species determined to date (Tab. 2).

Tab. 2 Mitogenomes of the nymphalids used in this study and their partial characteristics
To evaluate the degree of base bias for theP. sulpitiamitogenome, base-skewness was also measured in this study. The results showed that AT and GC-skewness values of the whole genome (measured from the major strand) were -0.048 and -0.178, respectively. This indicated that T and C were more frequently used than A and G in the genome, similar to results found in other nymphalid species used in this study (Tab. 3). However, when the two skewness values were considered separately, it was clear that the AT skew was the highest and the GC skew was the lowest of all the nymphalids in this study.

Tab. 3 Nucleotide composition and skewness of the nymphalid mitogenomes
3.2 Protein-coding genes
Twelve PCGs ofP. sulpitiamitogenome were initiated by typical ATN codons, except for thecox1gene. For theP. sulpitiaCOI gene, no typical ATN initiator was found in its starting region or in its neighboringtrnYsequences. As for thecox1initiation codon in animals, significantly different cases have been reported, for example, tetranucleotides such as TTAG inCoreana raphaelis(Kim et al, 2006), ATAA inDrosophila yakuba(Clary & Wolstenholme, 1985) are used, while hexanucleotides such as TATTAG inOstrinia nubilalisandOstrinia furnicalis(Coates et al, 2005), TTTTAG inBombyx mori(Yukuhiro et al, 2002), TATCTA inPenaeus monodon(Wilson et al, 2000), ATTTAA inAnopheles gambiae(Beard et al, 1993),Anopheles quadrimaculatus(Mitchell et al, 1993), andCeratitis capitata(Spanos et al, 2000) are used. Generally, the trinucleotide TTG was assumed to be thecox1start codon for some invertebrate taxa including insect species, such asPyrocoelia rufa(Bae et al, 2004),Caligula boisdnvalii(Hong et al, 2008), andAcraea issoria(Hu et al, 2010). In this study, however, according to sequence homologies with other available relevant insect species, the codon CGA was hypothesized to be thecox1initiator synapomorphically characteristic of most lepidopteran species (Kim et al, 2009, 2010).
Thenad4gene ofP. sulpitiaharbored a single T, rather than the common stop codon TAA. Incomplete termination codons are frequently observed in most insect mitogenomes including all the sequenced lepidopteran insects to date (Kim et al, 2009), which has been interpreted in terms of post-transcriptional polyadenylation, in which two A residues are added to create the TAA terminator (Anderson et al, 1981; Ojala et al, 1981).
The value of A+T content for all PCGs was 80.6%, whereas, the corresponding values for the major and minor strands were 79.2% and 83.1%, respectively. Both values were the highest of all the nymphalids analysed in this study (Tab. 4). Furthermore, the A+T content of the PCG third codon position was calculated to be 96.7%, which was significantly higher than those of the first (74.8%) and the second (70.5%) codon positions. This value was the highest of all the corresponding values among the nymphalids (Tab. 4). With regard to AT-skew, the degree of A+T bias was calculated in different strands of theP. sulpitiamitogenome PCGs: the major strand evidenced a value of -0.172, whereas the minor strand exhibited a value of -0.154. In contrast, for the GC-skew, the major and minor strands showed values of -0.100 and 0.266, respectively (Tab. 3). Additionally, the A+T bias of the PCG codon usage for theP. sulpitiamitogenome (the relative synonymous codon frequencies, RSCU) revealed that codons harboring A or T in the third position were frequently used compared to other synonymous codons (Tab. 5).

Tab. 4 Summary of base composition at each codon* position of the 13 PCGs in the nymphalid mitogenomes used in this study
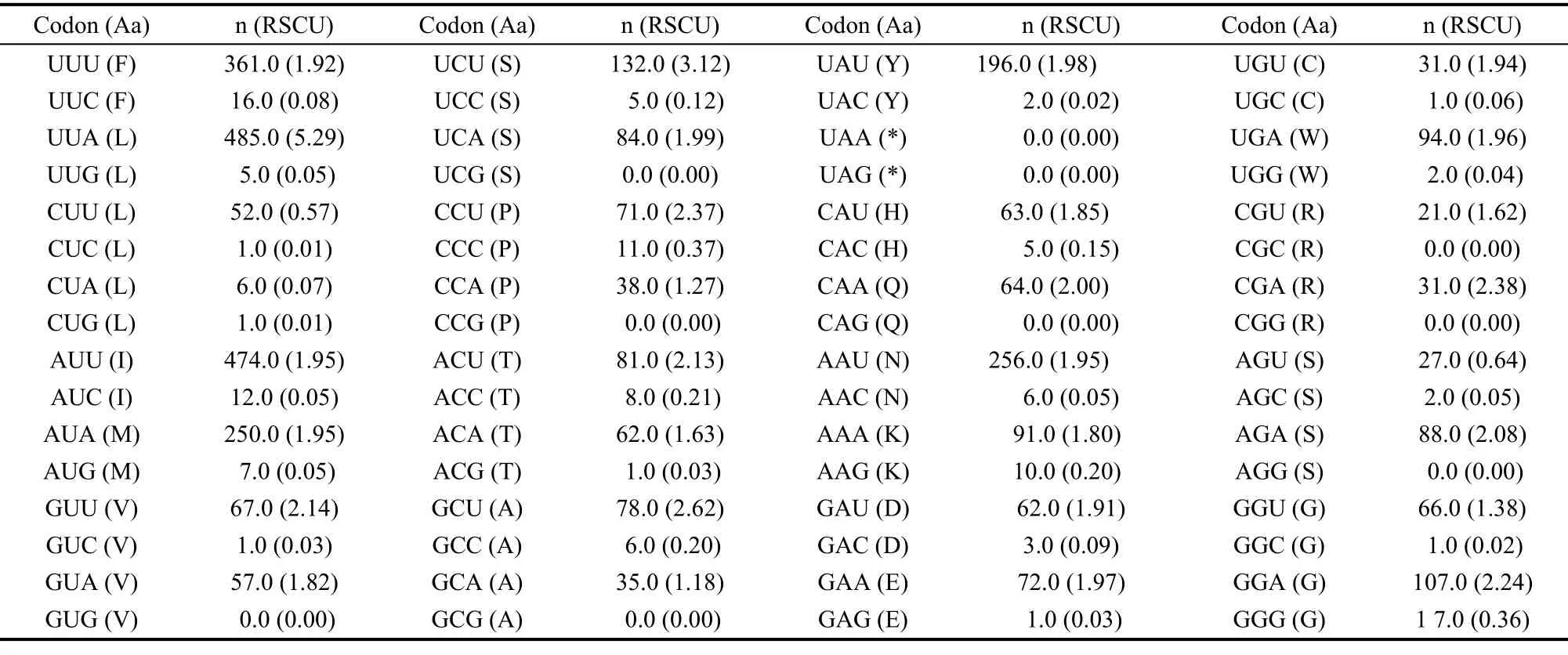
Tab. 5 Codon usage of the protein coding genes of the Parathyma sulpitia mitogenome
3.3 Transfer RNA and ribosomal RNA genes
TheP. sulpitiamitogenome harbored 22 tRNA genes, which were scattered throughout its whole region as is typically observed in metazoans including insects (Cha et al, 2007; Crozier & Crozier, 1993; Hong et al, 2008; Kim et al, 2010; Wilson et al, 2000; Yukuhiro et al, 2002). All tRNAs presented typical clover-leaf structure, with the unique exception oftrnS1(AGN), which lacked the dihydrouridine (DHU) stem (Fig. 2). TheP. sulpitiatRNAs harbored a total of 22 pair mismatches in their stems, with the number of mismatches inP. sulpitiaroughly the same as those detected in other lepidopteran species such asAntheraea pernyi(Liu et al, 2008) andEriogyna pyretorum(Jiang et al, 2009), but less than those inOchrogaster lunifer(Salvato et al, 2008). These tRNAs mismatches can be corrected through RNA-editing mechanisms, which are well known for arthropod mtDNA (Lavrov et al, 2000).
As in all other insect mitogenome sequences, two rRNA genes (rrnLandrrnS) were detected inP. sulpitia. They were located betweentrnL1(CUN) andtrnV, and betweentrnVand the A+T region, respectively (Fig. 1). The length of therrnLwas determined to be 1 319 bp, which was within the size range observed in the other available sequenced insects, from 470 bp inBemisia tabaci(Thao et al, 2004) to 1 426 bp inHyphantria cunea(Liao et al, 2010). The length of therrnSwas determined to be 779 bp, which was well within the size range observed in other completely sequenced insects, from 434 bp inOstrinia nubilalis(Clary & Wolstenholme, 1985) to 827 bp inLocusta migratoria(Flook et al, 1995).
3.4 Intergenic spacers and overlapping regions
The mtDNA genome ofP. sulpitiaincluded a total of 213 bp intergenic spacer sequences which were spread over 11 regions ranging in size from one to 121 bp. The largest spacer sequence (121 bp) was located between thetrnS1(AGN) and thetrnE, rather than between thetrnQand thenad2gene as found in other lepidopteran mitogenomes (Tab. 1). This spacer contained the highest A+T nucleotide (100%) of all the corresponding regions in all other lepidopterans determined. The sequence alignment of this spacer with partial A+T-rich region revealed a sequence homology of 74.4% (Fig. 3), suggesting that this spacer may have originated from a partial duplication of the A+T-rich region.

Fig. 3 Alignment of the largest spacer located between trnS1(AGN) and trnE and the partial A+T region
The second largest intergenic spacer was 52 bp long, located between thetrnQandnad2genes. This spacer is present in all lepidopteran mitogenomes sequenced, but absent in all non-lepidopteran insects (Hong et al, 2008). The sequence alignment of this spacer with the neighboringnad2gene revealed a sequence homology of 62%, and thus, this spacer was proposed to have been originated from a partial duplication of thenad2gene (Kim et al, 2009), with similar cases presented in other sequenced lepidopterans, such asArtogeia melete(70%) (Hong et al, 2009),C. raphaelis(62%) (Kim et al, 2006),Parnassius bremeri(70%) (Kim et al, 2009), andPhthonandria atrilineata(70%) (Yang et al, 2009). The other nine smaller intergenic spacers ranged in size from one to 11 bp were dispersed throughout the whole genome, and their details are listed in Tab. 1.
A total of 92 bp were identified as overlapping sequences varying from one to 35 bp in 15 regions of the genome (Tab. 2). The longest overlap was 35 bp located between thecox2andtrnKgenes, and the second largest was 20 bp long located betweentrnFandnad5. The third longest was 8 bp betweentrnWandtrnC, with similarly sized overlaps also detected in other lepidopteran species (Hong et al, 2008). As expected, the 7 bp overlap within theatp8andatp6reading frames, which is characteristic of many animal mitogenomes (Boore, 1999; Hong et al, 2008), was also detected in this study. In addition, a 5 bp and a 3 bp overlap were located betweencox1andtrnL(UUR), and betweentrnIandtrnQ, respectively. As for the remaining nine overlaps of 1 or 2 bp in size, their detailed cases are shown in Tab. 1.
3.5 A+T-rich region
The A+T-rich region ofP. sulpitiawas 349 bp in size, located betweenrrnSandtrnM(Fig. 1). This region showed the second highest A+T content (94.6%), slightly lower than the largest intergenic spacer (100%). This region included the ON(origin of minority or light strand replication), which was identified by the motif ATAGA located 20 bp downstream fromrrnS. Additionally, a motif ATAGA followed by 19 bp poly-T, which has been suggested as the structural signal for the recognition of proteins in the replication initiation of minor-strand mtDNA, was detected, which is similar to that observed in other lepidopteran species such as theBombyx mori(Yukuhiro et al, 2002). Finally, a few of multiple short microsatellite-like repeat regions, such as the (AT)7located 195 bp upstream fromrnnSand preceded by the ATTTA motif, were present, which was as expected as they are also detected in the majority of other sequenced lepidopterans (Hong et al, 2008; Hu et al, 2010; Kim et al, 2009; Mao et al, 2010; Pan et al, 2008; Wang et al, 2011; Xia et al, 2011). As for the tRNA-like sequences and the tandemly repeated elements often reported in other lepidopteran species (Kim et al, 2009; Pan et al, 2008), no relevant structures were detected in theP. sulpitiaA+T-rich region.
3.6 Phylogenetic analysis
An up-to-date and comprehensive classification of Nymphalidae was made by Ackery et al (1999) based on morphological characters, while work on molecular systematics of various lineages within Nymphalidae is beginning to clarify their relationships with interesting results (Brower et al, 2000; Wahlberg et al, 2003, 2005). Though the twelve subgroups of Nymphalidae (Libytheinae, Danainae, Charaxinae, Morphinae, Satyrinae, Calinaginae, Heliconiinae, Limenitidinae, Cyrestinae, Biblidinae, Apaturinae, and Nymphalinae) are widely accepted at the subfamily level, some relationships within this group remain unresolved. For example, the phylogenetic positions of Danainae, Libytheinae, and Limenitidinae within Nymphalidae are still controversial.
As for the Limenitidinae, its sister group within the Nymphalidae has been the subject of substantial debate (Freitas & Brown, 2004; Harvey, 1991). From a morphological view, the close relationships of Limenitidinae, Heliconiinae, Nymphalinae, and Apaturinae have never been suggested (de Jong et al, 1996; Freitas & Brown, 2004; Harvey, 1991). For example, Freitas & Brown (2004) conducted a cladistic analysis of Nymphalidae based on immature and adult morphological characters, and the results showed that Limenitidinae is sister to the grouping of (Apaturinae + (Calinaginae + Satyrinae)), exclusive of the remaining nymphalidae taxa (Freitas & Brown, 2004). However, phylogenetic analyses based on molecular sequence data have convincingly suggested that Limenitidinae is the sister group of Heliconiinae (Brower, 2000; Wahlberg et al, 2003, 2005; Zhang et al, 2008). In this study, the ML and BI phylogenetic analyses based on the mitogenomic data of the nine available nymphalids, including that ofP. sulpitiaand other unpublished species, revealed the following relationships: (Danainae + ((Libytheinae + ((Satyrinae + Calinaginae) + (Apaturinae + (Heliconiinae + Limenitidinae) + Nymphalinae))))) with high support values (Fig. 4), which is congruent with those reported by Wahlberg et al (2003, 2005) and Brower (2000).

Fig. 4 ML (A) and BI (B) trees of the nymphalid species based on nucleotide sequences of the 13 protein-coding genes Numbers at nodes are bootstrap values/posterior probabilities.
In conclusion, the complete mitogenome ofP. sulpitiaharbored nearly the same characters as those of other nymphalids. Phylogenetic analysis on a mitogenomic level indicated that Limenitidinae was most closely related to Heliconiinae than other groups of Nymphalidae in this study, strongly supporting the results of former molecular studies, while contradicting the prevailing speculations based on morphological characters.
Abascal F, Posada D, Zardoya R. 2007. MtArt: a new model of amino acid replacement for arthropoda[J].Mol Biol Evol,24(1): 1-5.
Ackery PR. 1984. Systematic and faunistic studies on butterflies [M] //Vane-Wright RI, Ackery PR. (Eds.), Systematic and Faunistic Studies on Butterflies. Princeton, USA: Princeton University Press, 9-21.
Ackery PR, de Jong R, Vane-Wright RI. 1999. The butterflies: Hedyloidea, Hesperoidea, and Papilionoidea [M] //Kristensen NP. (Ed.), Lepidoptera, Moths and Butterflies. Handbook of Zoology, Lepidoptera. Berlin: De Gruyter, 263-300.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool [J].J Mol Biol,215(3): 403-410.
Anderson S, Bankier AT, Barrell BG, de Bruijin MHL, Coulson AR, Droujn J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJH, Staden R, Young IG. 1981. Sequence and organization of the human mitochondrial genome [J].Nature,290(5806): 457-465.
Bae JS, Kim I, Sohn HD, Jin BR. 2004. The mitochondrial genome of the firefly,Pyrocoelia rufa: complete DNA sequence, genome organization, and phylogenetic analysis with other insects [J].Mol Phylogenet Evol,32(3): 978-985.
Beard CB, Hamm DM, Colllins FH. 1993. The mitochondrial genome of the mosquitoAnopheles gambiae: DNA sequence, genome organization, and comparisons with mitochondrial sequences of other insects [J].Insect Mol Biol,2(2): 103-124.
Boore JL. 1999. Animal mitochondrial genomes [J].Nucleic Acids Res,27(8): 1767-1780.
Brower AVZ. 2000. Phylogenetic relationships among the Nymphalidae (Lepidoptera) inferred from partial sequences of thewinglessgene [J].Proc R Soc Lond B,267(1449): 1201-1211.
Cameron SL, Whiting MF. 2008. The complete mitochondrial genome of the tobacco hornworm,Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths [J].Gene,408(1-2): 112-123.
Cha SY, Yoon HJ, Lee EM, Yoon MH, Hwang JS, Jin BR, Han YS, Kim I. 2007. The complete nucleotide sequence and gene organization of the mitochondrial genome of the bumblebee,Bombus ignitus(Hymenoptera: Apidae) [J].Gene,392(1-2): 206-220.
Clary DO, Wolstenholme DR. 1985. The mitochondrial DNA molecule ofDrosophila yakuba: nucleotide sequence, gene organization, and genetic code [J].J Mol Evol,22(3): 252-271.
Clayton DA. 1992. Transcription and replication of animal mitochondrial DNA [J].Int Rev Cytol,141: 217-232.
Coates BS, Sumerford DV, Hellmich RL, Lewis LC. 2005. Partial mitochondrial genome sequences ofOstrinia nubilalisandOstrinia furnicalis[J].Int J Biol Sci,1(1): 13-18.
Crozier RH, Crozier YC. 1993. The mitochondrial genome of the honeybeeApis mellifera: complete sequence and genome organization [J].Genetics,133(1): 97-117.
de Jong R, Vane-Wright RI, Ackery PR. 1996. The higher classification of butterflies (Lepidoptera): problems and prospects [J].Entomol Scand,27(1): 65-101.
Ehrlich PR. 1958. The comparative morphology, phylogeny and higher classification of the butterflies (Lepidoptera) [J].Syst Entomol,10: 11-32.
Fiedler K. 2010. The coming and going of Batesian mimicry in a Holarctic butterfly clade.BMC Biol, 8(1): 122.
Flook PK, Rowell CHF, Grellissen G. 1995. The sequence organisation, and evolution of theLouocsta migratoriamitochondrial genome[J].J Mol Evol,41(6): 928-941.
Freitas AVL, Brown KS Jr. 2004. Phylogeny of the Nymphalidae (Lepidoptera) [J].Syst Biol,53(3):363-383.
Hao JS, Li CX, Sun XY, Yang Q. 2005. Phylogeny and divergence time estimation of cheilostome bryozoans based on mitochodrial 16S rRNA sequences [J].Chn Sci Bull,50(12): 1205-1211.
Harvey DJ. 1991. Higher classification of the Nymphalidae, Appendix B [M] //Nijhout HF. (Ed.), The Development and Evolution of Butterfly Wing Patterns. Washington, DC: Smithsonian Institution Press, 255-273.
Hong GY, Jiang ST, Yu M, Yang Y, Li F, Xue FS, Wei ZJ. 2009. The complete nucleotide sequence of the mitochondrial genome of the cabbage butterfly,Artogeia melete(Lepidoptera: Pieridae) [J].Acta Biochim Biophys Sin,41(6): 446-455.
Hong MY, Lee EM, Jo YH, Park HC, Kim SR, Huang JS, Jin BR, Kang PD, Kim KG, Han YS, Kim I. 2008. Complete nucleotide sequence and organization of the mitogenome of the silk mothCaligula boisduvalii(Lepidoptera: Saturniidae) and comparison with other lepidopteran insects [J].Gene,413(1-2): 49-57.
Hu J, Zhang DX, Hao JS, Huang DY, Cameron S, Zhu CD. 2010. The complete mitochondrial genome of the yellow coaster,Acraea issoria(Lepidoptera: Nymphalidae: Heliconiinae: Acraeini): sequence, gene organization and a unique tRNA translocation event [J].Mol Biol Rep,37(7): 3431-3438.
Jiang ST, Hong GY, Yu M, Li N, Yang Y, Liu YQ, Wei ZJ. 2009. Characterization of the complete mitochondrial genome of the giant silkworm moth,Eriogyna pyretorum(Lepidoptera: Saturniidae) [J].Int J Biol Sci,5(4): 351-365.
Kim I, Lee EM, Seol KY, Yun EY, Lee YB, Hwang JS, Jin BR. 2006. The mitochondrial genome of the Korean hairstreak,Coreana raphaelis(Lepidoptera: Lycaenidae) [J].Insect Mol Biol,15(2): 217-225.
Kim MI, Beak JY, Kim MJ, Jeong HC, Kim KG, Bae CH, Han YS, Jin BR, Kim I. 2009. Complete nucleotide sequence and organization of the mitogenome of the red-spotted Apollo butterfly,Parnassius bremeri(Lepidoptera: Papilionidae) and comparison with other lepidopteran insects [J].Mol Cell,28(4): 347-363.
Kim MJ, Wan XL, Kim KG, Hwang JS, Kim I. 2010. Complete nucleotide sequence and organization of the mitogenome of endangeredEumenis autonoe(Lepidoptera: Nymphalidae) [J].Afr J Biotechnol,9(5): 735-754.
Lavrov DV, Brown WM, Boore JL. 2000. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipedeLithobius forficatus[J].Proc Natl Acad Sci USA,97(25): 13738-13742.
Liao F, Wang L, Wu S, Li YP, Zhao L, Huang GM, Niu CJ, Liu YQ, Li MG. 2010. The complete mitochondrial genome of the fall webworm,Hyphantria cunea(Lepidoptera: Arctiidae) [J].Int J Biol Sci,6(2): 172-186.
Liu Y, Li Y, Pan M, Dai F, Zhu X, Lu C, Xiang Z. 2008. The complete mitochondrial genome of the Chinese oak silkmoth,Antheraea pernyi(Lepidoptera: Saturniidae) [J].Acta Biochim Biophys Sin,40(8): 693-703.
Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence [J].Nucleic Acids Res,25(5): 955-964.
Mao ZH, Hao JS, Zhu GP, Hu J, Si MM, Zhu CD. 2010. Sequencing and analysis of the complete mitochondrial genome ofPieris rapaeLinnaeus (Lepidoptera: Pieridae) [J].Acta Entomol Sin,53(11): 1295-1304.
Mitchell SE, Cockburn AF, Seawright JA. 1993. The mitochondrial genome ofAnopheles quadrimaculatusspecies A: complete nucleotide sequence and gene organization [J].Genome,36(6): 1058-1073.
Ojala D, Montoya J, Attardi G. 1981. tRNA punctuation model of RNA processing in human mitochondria [J].Nature,290(5806): 470-474.
Pan MH, Yu QY, Xia YL, Dai FY, Liu YQ, Lu C, Zhang Z, Xiang ZH. 2008. Characterization of mitochondrial genome of Chinese wild mulberry silkworm,Bomyx mandarina(Lepidoptera: Bombycidae) [J].Sci Chn: Ser C-Life Sci,51(8): 693-701.
Platt AP, Maudsley JR. 1994. Continued interspecific hybridization betweenLimenitis(Basilarchia)arthemis astyanaxandL.(B.)archippusin the southeastern US (Nymphalidae) [J].J Lepidopt Soc,48(3): 190-198.
Posa D, Krandall KA. 1998. Modeltest: testing the model of DNA substitution [J].Bioinformatics,14(9): 817-818.
Ronquist F, Huelsenbeck JP. 2003. MRBAYES 3: Bayesian phylogenetic inference under mixed models [J].Bioinformatics,19(12): 1572-1574.
Salvato P, Simonato M, Battisti A, Negrisolo E. 2008. The complete mitochondrial genome of the bag-shelter mothOchrogaster lunifer(Lepidoptera, Notodontidae) [J].BMC Genomics,9: 331.
Simon C, Frati F, Bekenbach A, Crespi B, Liu H, Flook P. 1994. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers [J].Ann Entomol Soc Am,87(6): 651-701.
Singh VK, Mangalam AK, Dwivedi S, Naik S. 1998. Primer premier: Program for design of degenerate primers from a protein sequence [J].Biotechniques,24(2): 318-319.
Spanos L, Koutroumbras G, Kotsyfakis M, Louis C. 2000. The mitochondrial genome of the Mediterranean fruit fly,Ceratitis capitata[J].Insect Mol Biol,9(2):139-144.
Swofford DL. 2002. PAUP*: Phylogenetic analysis using parsimony (* and other methods), version 4.0 [M]. Sunderland: Sinauer Associates.
Thao ML, Baumann L, Baumann P. 2004. Organization of the mitochondrial genomes of whiteflies, aphids, and psyllids (Hemiptera, Sternorrhyncha) [J].BMC Evol Biol,4: 25.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The Clustal X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools [J].Nucl Acids Res,25(24): 4876-4882.
Wahlberg N, Braby M F, Brower AVZ, de Jong R, Lee MM, Nylin S, Pierce NF, Sperling FAH, Vila R, Warren AD, Zakharov E. 2005. Synergistic effects of combining morphological and molecular data in resolving the phylogeny of butterflies and skippers [J].Proc R Soc Lond B,272(1572): 1577-1586.
Wahlberg N, Weingartner E, Nylin S. 2003. Towards a better understanding of the higher systematics of Nymphalidae (Lepidoptera: Papilionoidea) [J].Mol Phylogenet Evol,28(3): 473-484.
Wahlberg N, Wheat CW. 2008. Genomic outposts serve the phylogenomic pioneers: designing novel nuclear markers for genomic DNA extractions of lepidoptera [J].Syst Biol,57(2): 231-242.
Wang XC, Sun XY, Sun QQ, Zhang DX, Hu J, Yang Q, Hao JS. 2011. Complete mitochondrial genome of the laced fritillaryArgyreus hyperbius(Lepidoptera: Nymphalidae) [J].Zool Res,32(5): 465-475.
Wilson K, Cahill V, Ballment E, Benzie J. 2000. The complete sequence of the mitochondrial genome of the crustaceanPenaeus monodon: are Malacostracan crustaceans more closely related to insects than to Branchiopods [J].Mol Biol Evol,17(6): 863-874.
Wolstenholme DR. 1992. Animal mitochondrial DNA: structure and evolution [J].Int Rev Cytol,141: 173-216.
Wu LW, Lees DC, Yen SH, Lu CC, Hsu YF. 2010. The complete mitochondrial genome of the near-threatened swallowtail,Agehana maraho(Lepidoptera: Papilionidae): evaluating sequence variability and suitable markers for conservation genetic studies [J].Entomol News,121(3): 267-280.
Xia J, Hu J, Zhu GP, Zhu CD, Hao JS. 2011. Sequencing and analysis of the complete mitochondrial genome ofCalinaga davidisOberthür (Lepidoptera: Nymphalidae) [J].Acta Entomol Sin,54(5): 555-565.
Xia X, Xie Z. 2001. DAMBE: Data analysis in molecular biology and evolution [J].J Hered,92(4): 371-373.
Yang L, Wei ZJ, Hong GY, Jiang ST, Wen LP. 2009. The complete nucleotide sequence of the mitochondrial genome ofPhthonandria atrilineata(Lepidoptera: Geometridae) [J].Mol Biol Rep,36(6): 1441-1449.
Yang Z, Rannala B. 1997. Bayesian phylogenetic inference using DNA sequences: A Markov chain Monte Carlo method [J].Mol Biol Evol,14(7): 717-724.
Yukuhiro K, Sezutsu H, Itoh M, Shimizu K, Banno Y. 2002. Significant levels of sequence divergence and gene rearrangements have occurred between the mitochondrial genomes of the wild mulberry silkmoth,Bombyx mandarina, and its close relative, the domesticated silkmoth,Bombyx mori[J].Mol Biol Evol,19(8): 1385-1389.
Zhang M, Zhong Y, Cao TW, Geng YP, Zhang Y, Jin K, Ren ZM, Zhang R, Guo YP, Ma EB. 2008. Phylogenetic relationship and morphological evolution in the subfamily Limenitidinae (Lepidoptera: Nymphalidae) [J].Prog Nat Sci,18(11): 1357-1364.
残锷线蛱蝶线粒体基因组全序列及其系统学意义
田丽丽1, 孙晓燕2, 陈 梅1, 盖永华2, 郝家胜1,2,*, 杨 群2,*
(1. 安徽师范大学 生命科学学院分子进化与生物多样性研究室, 安徽 芜湖 241000; 2. 中国科学院南京地质古生物研究所 现代古生物学与地层学国家重点实验室, 南京 210008)
对残锷线蛱蝶(Parathyma sulpitia)(鳞翅目:蛱蝶科)线粒体基因组全序列进行了测定。结果表明:残锷线蛱蝶线粒体基因组全序列全长为15 268 bp, 除了在trnS1(AGN) 和trnE基因之间有一段121 bp长的基因间隔外, 其基因的排列顺序及排列方向与大多数已测鳞翅目物种基本一致。在蛋白质编码基因中, 除cox1以CGA作为其起始密码子之外, 其余12个蛋白质编码基因都以标准的ATN作为起始密码子。此外, 除nad4基因以单独的T为终止密码子, 其余12个蛋白质编码基因都以TAA结尾。除trnS1(AGN) 缺少DHU臂之外, 22个tRNA基因都显示典型的三叶草形二级结构。除A+T富集区外的非编码序列中, 线粒体基因组共含有11个基因间隔区。其中,最长的一个121 bp的基因间隔区位于trnS1(AGN)和trnE之间, 其A+T含量高达100%。另外, 和其他鳞翅目物种一样, 在其A+T富集区的3'端有一段长达18 bp的poly-T结构。A+T富集区内部没有明显的小卫星样多拷贝重复序列, 而含有一些微卫星样的重复结构。本研究基于 13种蛋白编码基因序列的组合数据, 用最大似然法和贝叶斯法对蛱蝶科几个主要亚科间共 9个代表物种间的系统发生关系进行了分析。结果表明, 本研究的结果与前人的分子系统学研究结论基本吻合(其中, 线蛱蝶亚科和釉蛱蝶亚科互为姐妹群), 而与形态学的研究结论不一致。
2011-11-18;接受日期:2012-02-28
残锷线蛱蝶; 鳞翅目; 蛱蝶科; 线蛱蝶亚科; 线粒体基因组
Q969.42; Q969.439.2 ; Q754
A
0254-5853-(2012)02-0133-11
10.3724/SP.J.1141.2012.02133
date:2011-11-18; < class="emphasis_bold">Accepted date
date: 2012-02-28
s:This work was supported by the National Natural Science Foundation of China (41172004), the CAS/SAFEA International Partnership Program for Creative Research Teams, Chinese Academy of Sciences (KZCX22YW2JC104), the Provincial Key Project of the Natural Science Foundation from the Anhui Province, China (KJ2010A142), and the Open Funds from the State Key Laboratory of Palaeobiology and Stratigraphy, Nanjing Institute of Geology and Palaeontology, Chinese Academy of Sciences* Corresponding authors (通信作者), E-mail: jshaonigpas@sina.com; qunyang@nigpas.ac.cn
猜你喜欢
幼儿100(2020年25期)2020-10-22
生物学通报(2020年11期)2020-10-22
生物学通报(2020年11期)2020-10-22
武夷科学(2019年2期)2019-12-20
发明与创新·中学生(2019年6期)2019-06-26
西藏人文地理(2019年6期)2019-02-21
中成药(2018年7期)2018-08-04
小学生必读(低年级版)(2018年4期)2018-08-01
中成药(2018年3期)2018-05-07
中国烟草学报(2012年2期)2012-04-09

- Zoological Research的其它文章
- 杭州城市环境中白头鹎的繁殖生态