
黏着斑激酶及其小分子抑制剂作为抗肿瘤药物的研究进展
2014-03-08 02:07杨超王重庆陈颖田巍朱驹
药学进展 2014年9期
杨超,王重庆,陈颖,田巍,朱驹
(第二军医大学药学院药物化学教研室,上海 200433)
黏着斑激酶及其小分子抑制剂作为抗肿瘤药物的研究进展
杨超,王重庆,陈颖,田巍,朱驹*
(第二军医大学药学院药物化学教研室,上海 200433)
抗黏着斑激酶是一种非受体型酪氨酸蛋白激酶,在许多肿瘤的发生和发展过程中均有过表达。研究表明,作为细胞内重要的骨架蛋白和调节多种细胞信号通路的关键分子,黏着斑激酶在肿瘤发生、发展、迁移和侵袭的各个阶段都起着重要作用。因此,以黏着斑激酶作为抗肿瘤靶点开发其抑制剂的研究受到广泛关注。综述黏着斑激酶的结构与功能、它与肿瘤的关联及其小分子抑制剂的研究与开发。
黏着斑激酶;细胞信号通路;靶点;黏着斑激酶小分子抑制剂;抗肿瘤活性
黏着斑激酶(focaladhesionkinase,FAK)是一种位于黏着斑、由1028个氨基酸组成的细胞质酪氨酸激酶和骨架蛋白,参与多种受体或非受体酪氨酸激酶信号通路,包括肿瘤蛋白Src、血管内皮生长因子受体-3(VEGFR-3)、p53、磷脂酰肌醇-3激酶(PI3K)和胰岛素样生长因子-1(IGF-1)等通路,对细胞的存活、增殖、迁移和侵袭等生物行为都有调节作用。已有研究表明,在许多肿瘤细胞中都有FAK的过表达。因此,FAK已成为研究与开发新型抗肿瘤药物的重要靶标。
1 黏着斑激酶的结构与功能
FAK蛋白分子内按功能大致可划分为:N端的FERM区、脯氨酸丰富区(Pro1、Pro2和Pro3)、中间的激酶区和C端的黏着斑靶向区(focaladhesiontarget,FAT)(见图1)[1]。其中,N端的FERM区可直接结合胞内的整合素β1亚基单元[2](又见:Schaller等,J Cell Biol,1995年)和一些细胞膜受体[如表皮生长因子受体(EGFR)、血小板衍生生长因子受体(PDGFR)和c-Met受体][3](又见:Sieg等,Nat Cell Biol,2000年;Golubovskaya等,J Biol Chem,2002年),FERM具有三叶草形结构,调节着FAK的催化活性,有研究发现截短FERM区的FAK分子磷酸化活性和催化活性均提高,表明FERM对FAK起负性调节作用,而FERM区与激酶区的结合使得FAK维持在一种失活构象;且FERM能与p53分子相互作用,促进肿瘤细胞的存活,当FAK分子向细胞核内转运后,FERM的F1亚结构域与p53结合,F2亚结构域则引起p53向细胞核转运,F3亚结构域通过与Mdm-2分子相互作用而导致p53降解[4]。FAK的激酶区也称催化区,有着高度保守的氨基酸序列,可使PI3K、生长因子受体结合蛋白2(GRB2)、Cas和Src等蛋白中相应的氨基酸残基磷酸化。
Pro1位于FERM区和激酶区之间,有着与SH3结构域结合的位点,可与Src等蛋白结合。靠近C端的Pro2与Pro3可介导FAK与含有SH3结构域的蛋白分子间的相互作用,包括接头蛋白p130Cas、Graf(包括Rho-A特异性GTPase激活蛋白和FAK相关的鸟苷酸三磷酸调节激酶)和ASAP1(ARF-GAP含有SH3结构域、ANK重复序列和PH区域)(Harte等,J Biol Chem,1996年;Taylor等,J Biol Chem,1998年;Liu等,Mol Biol Cell,2002年)。
C端的FAT是FAK黏附到黏着斑的功能域,包含黏着相关蛋白[如桩蛋白(paxillin)和踝蛋白(talin)]结合位点,这些蛋白可直接结合于整合素在细胞质的区域,还可结合FAT,从而介导黏着复合物的形成。
FAK有6个可被磷酸化的酪氨酸位点:Y397、Y407、Y576、Y577、Y861和Y925,这些磷酸化位点都是FAK发挥信号转导的关键部位,其中Y397和Y407位于N端,Y576和Y577位于激酶区的活化环内,Y861和Y925位于C端。Y397是FAK的一个自磷酸化位点,为Src家族蛋白提供了一个高亲和力的结合位点,对下游信号通路起至关重要的作用。非受体型蛋白激酶家族同样包括两个与FAK相关的成员:FAK相关非激酶(FRNK)和富含脯氨酸的酪氨酸激酶2(Pyk2),FRNK是FAKC端基因单独表达的一个p41/43蛋白,可与FAK竞争性结合黏着位点,为FAK的一种内源性抑制剂;Pyk2与FAK也有同源性,但它并不在所有细胞类型中表达,FAK基因敲除实验显示,Pyk2有部分补偿FAK缺失的潜能[1]。
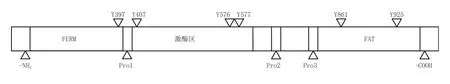
图1 FAK分子结构示意图Figure 1 Molecular structure diagram of FAK
2 黏着斑激酶与肿瘤
在肿瘤形成的过程中,肿瘤细胞会发生形态学和结构的改变,细胞表面黏附性降低,流动性增大,大量的血管生成以供应营养,从而使肿瘤细胞得以转移、侵袭及增殖。FAK过表达于多种癌细胞中,包括脑癌、卵巢癌、结肠癌、乳腺癌、前列腺癌、肝癌和甲状腺癌等,而且其过表达与这些肿瘤的侵袭能力密切相关[5]。实验研究显示,通过过表达显性失活的FAK片段,可抑制FAK信号通路,减少胶质母细胞瘤和卵巢癌细胞的侵袭[6]。大量研究表明,FAK磷酸化后参与细胞的多种信号通路,从而影响细胞的增殖、生存、迁移和侵袭等多种功能(见图2)[7],其中FAK的Y397位点磷酸化可诱导Src蛋白结合到Pro1上,继而引起Y576、Y577和Y925位点的磷酸化,致使FAK与桩蛋白和p130Cas的结合,导致细胞支架和形态学的改变。FAK可通过调节RhoGTP酶特别是RhoA、Rac-1和Cdc42来介导细胞能动性和附着力,并能通过Cas–Crk–DOCK–ELMO复合物激活Rac-1而上调板状伪足的形成[8](又见:Parsons,J Cell Sci,2003年)。此外,研究发现,FAK/AKT信号通路介导的基质金属蛋白酶-2(MMP-2)和MMP-9的激活可诱导癌细胞的迁移和侵袭[9],而FAK-siRNA在肝癌细胞SK-hep1和SMMC7721中能降低MMP-2和MMP-9的表达,从而影响癌细胞的侵袭和转移[10]。

图2 FAK磷酸化后参与的细胞信号通路示意图Figure 2 Diagram of FAK post-phosphorylation-involved cell signaling pathways
实验研究表明,FAK在神经胶质瘤样本中的表达水平远远超过在正常大脑中的水平,且在侵入性肿瘤周围FAK的表达增强,提示FAK在癌细胞的侵袭中有所作用[5];而且,FAK在人体神经胶质瘤活组织及其U251MG细胞异种移植模型的微血管内皮细胞中有表达,可见FAK与肿瘤的血管生成有关联(Haskell等,Clin Cancer Res,2003年);此外,野生型FAK在SF767和G112P神经胶质瘤中的过表达能促进肿瘤细胞的迁移、侵袭和增殖[11](又见:Lipinski等,Mol Cancer Res,2003年)。绝大多数胰腺癌病人的肿瘤组织中都存在FAK的过表达,而FAK抑制剂可通过多种途径降低胰腺癌细胞的生存力,抑制其生长,促进其凋亡[12-13],但FAK抑制剂对正常细胞仅有极少影响,且FAK可促进迁移细胞头部板状伪足的形成,从而促进细胞的迁移及癌症的扩散[14]。据报道,FAK的过表达在鳞状细胞癌中是一个常见的早期事件,不过也会出现在肿瘤的增殖发展期[15];FAK可通过提高细胞的流动性来增强鳞状细胞癌的侵袭性[16]。提示,FAK表达的急剧增加极有可能预示着癌症的扩散,FAK可作为抑制鳞状细胞癌细胞向其他组织侵袭和向淋巴系统扩散的靶标。
有研究显示,在卵巢癌细胞中,儿茶酚胺类药物可激活FAK的表达,从而抑制癌细胞的凋亡,促进癌细胞的增殖、侵袭和生长[17],且FAK的过表达可在应激源的刺激下(如激活PI3K-AKT通路或诱导细胞凋亡的化学疗法)保护细胞,故FAK的过表达可作为预测卵巢癌治疗效果的指标[6];FAK表达下调则可增强多西紫杉醇对卵巢癌的细胞毒性,且在体外实验中,抑制FAK的磷酸化,可减少卵巢癌细胞的迁移和侵袭[18];在卵巢癌体内实验中,采用干扰RNA技术来沉默FAK,可产生抗肿瘤和抗血管生成的效果,并增强化疗的疗效[19-20]。黑色素瘤小鼠模型实验显示,FAK的反义寡核苷酸类抑制剂可抑制肿瘤的生长和转移,但对正常组织几乎无影响,且抑制FAK的表达,能降低瘤体的平均质量;而体外实验显示,通过siRNA技术来破坏FAK的表达或通过FRNK过量表达来干扰FAK与整合素复合物(即黏着位点)的结合,可显著抑制B16F10黑色素瘤细胞的迁移能力[21]。乳腺癌AU-565细胞的体外实验显示,作为侵袭性乳腺癌预后指标的HER2可通过激活Src/FAK信号通路而影响乳腺癌细胞的迁移,而采用siRNA技术降低FAK的表达,则能削弱癌细胞的跨上皮迁移作用[22]。
3 黏着斑激酶抑制剂的研究与开发
目前,进入临床前或临床研究的FAK抑制剂几乎均为小分子抑制剂。由于FAK与许多细胞信号蛋白都有作用,因此依据作用机制的不同,作为FAK靶向抗肿瘤药物的FAK小分子抑制剂大致可分为两大类:ATP依赖型和ATP非依赖型。ATP依赖型FAK小分子抑制剂能干扰FAK催化区域的活性,可能会影响多个下游信号通路,造成较为广泛的副作用,而非ATP依赖型FAK小分子抑制剂如变构FAK抑制剂则可阻滞特定的蛋白质-蛋白质相互作用(如p53与FAK的相互作用),从而抑制FAK的活性。
3.1 ATP依赖型FAK小分子抑制剂
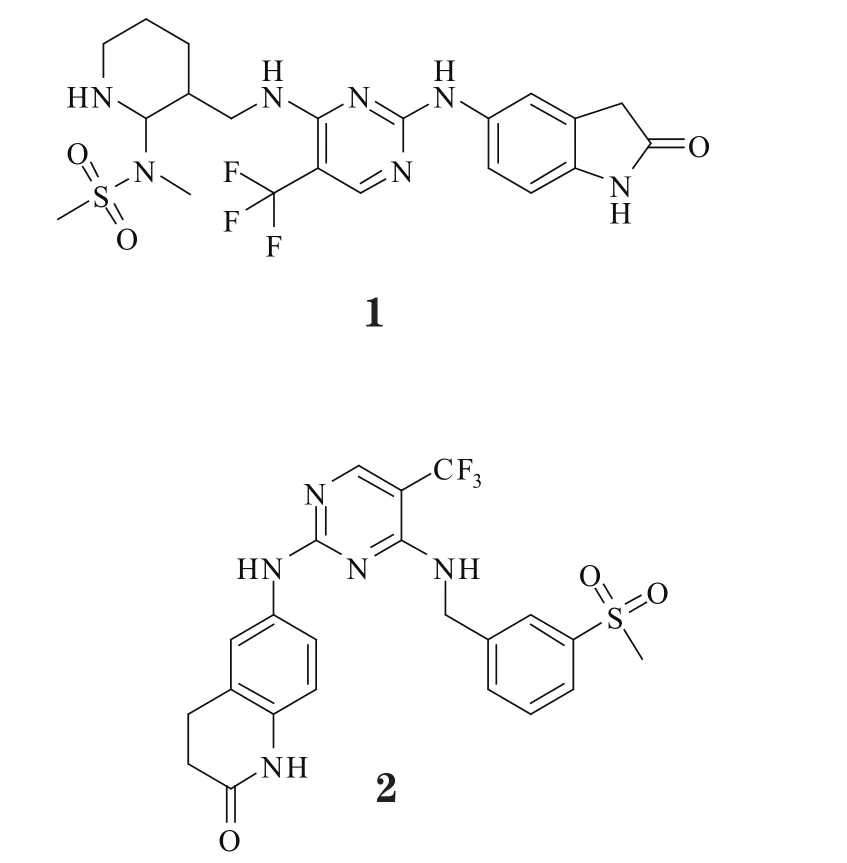
3.1.1 PF-562271PF-562271(1)是一种有效的ATP竞争性FAK/Pyk2可逆抑制剂,体内IC50为1.5/14nmol·L-1,而对FAK的体外抑制活性(IC50=5nmol·L-1)是对Pyk2的10倍,且较对其他非靶标激酶高100倍以上,表现出高度选择性;其在肿瘤模型小鼠体内抑制FAK磷酸化的EC50为93μg·L-1[23]。研究发现,该化合物可剂量依赖性地阻断FAK-Y397磷酸化,抑制肿瘤细胞、成纤维细胞和巨噬细胞的迁移[24];它在肿瘤模型小鼠体内可抑制肿瘤细胞的生长、侵袭和转移,但无致肿瘤坏死和细胞凋亡的作用,对血管生成也无作用[25]。另有研究显示,3.3μmol·L-1的PF-562271能导致前列腺癌PC3-M细胞的细胞周期停滞在G1期[23],且在鸡胚绒毛尿囊膜实验中,1nmol·L-1的PF-56227可强有效阻断碱性成纤维细胞生长因子(bFGF)刺激的血管生成,但其对血管渗漏无明显影[4];在肝脏实体瘤模型实验中,PF-562271和舒尼替尼的联合治疗可抑制血管发生和肿瘤增殖[26]。且研究表明,PF-562271是治疗胰腺导管癌和伴有骨质疏松症的癌症患者的一个潜在有效药物[24]。PF-562271用于治疗头颈癌、前列腺癌和胰腺癌的Ⅰ期临床研究始于2005年12月,2010年结束,证实其具有较低毒性和很强的肿瘤抑制作用[27]。
PF-562271是甲基磺酰胺二氨基嘧啶类似物,有着良好的物理化学性质,符合类药5原则。X射线晶体学分析表明,PF-562271结合在FAK的ATP结合位点,与激酶铰链区的主链上Cys-502形成两个氢键;与氨基相连的吲哚基团沿FAK活性部位铰链区主链往裂缝外延伸,其上氧原子与侧链Arg-426形成氢键;另一端的甲基磺酰胺二氨基嘧啶则从FAK活性部位向激活环区延伸,嘧啶基与Leu-567有疏水性相互作用,磺胺基上氧原子与Asp-564的NH有氢键作用。由于磺胺与高度保守蛋白激酶的可变的特殊激活环区能产生相互作用,故使得PF-562271对FAK有着很好的选择性[23]。
3.1.2 PF-573228PF-573228(2)是一种ATP竞争性FAK抑制剂,能靶向结合在激酶催化区域的ATP结合口袋,有效抑制重组FAK及内源性FAK的催化区域活性,对FAK/AKT通路有抑制活性但不影响胶原受体GPVI通路的活性[28]。本品体外抑制FAK催化片段纯化重组体的IC50为4nmol·L-1,比作用于Pyk2、细胞周期素依赖性蛋白激酶1/7(CDK1/7)和糖原合成酶激酶-3β(GSK-3β)的选择性高50~250倍。细胞水平的实验研究显示,PF-573228作用于REF52成纤维细胞、前列腺癌PC3细胞和卵巢癌SKOV-3细胞时,抑制FAK的Y-397磷酸化的IC50为30~500nmol·L-1;然而,1μmol·L-1的PF-573228可抑制80%FAK磷酸化且降低黏着斑转换,却不抑制细胞生长[25]。
3.1.3 TAE226TAE226(3)是经典的ATP竞争性酪氨酸激酶小分子抑制剂,对FAK和IGF-1受体(IGF-1R)具有相同抑制活性,IC50为100~300nmol·L-1。实验研究显示,TAE226可抑制FAK的Y-397和AKT的Ser-473磷酸化,致使人口腔鳞状细胞癌SAS细胞的肌纤蛋白结构发生改变,造成细胞间质的形态学改变,减少细胞黏合,有效抑制癌细胞增殖、迁移和侵袭,并导致由半胱天冬酶(Caspase)介导的细胞凋亡;且它能有效抑制肿瘤毛细血管的生成。提示,TAE226是治疗口腔鳞状细胞癌的一个非常有前景的先导化合物[29]。而且,TAE226在体内外实验中对一系列恶性肿瘤(如神经胶质瘤、卵巢癌、脑癌、乳腺癌、食管癌、胃肠道间质癌等)都表现出有效的抗增殖和抑瘤作用。如一项体外研究显示,本品能有效诱导过表达Scr和EGFR的乳腺癌细胞凋亡,其对FAK、胰岛素受体(InsR)和IGF-1R的IC50分别为5.5、44和140nmol·L-1[24]。
TAE226为二苯胺基嘧啶类化合物,可结合在FAK的ATP结合口袋,以一种特定的形式与激酶铰链区作用,其甲胺酰基中的羰基对诱导产生的DFG超二级结构的罕见螺旋构象起稳定作用,而这种螺旋构象在FAK与PF-562271形成的复合物中也有报道[23],它有助于提高化合物的选择性。TAE226-FAK的复合物晶体结构模型显示,TAE226嘧啶环上的N和嘧啶环与苯甲醚相连的N可与FAK的Cys502形成两个氢键,嘧啶环上的C则与FAK的Ala452和Leu553有疏水性相互作用,2-甲氧基苯胺上的C与FAK的Ile428和Gly505有相互作用,嘧啶环上C5位的氯伸入FAK的ATP结合口袋并位于Met499的侧面;TAE226苯胺环的结构几乎不与激酶蛋白作用,它的引入可改善化合物的药动学性质,邻位的甲胺甲酰基位于FAK激活区的DFG超二级结构的侧面,其上的羰基与DFG超二级结构上的Asp564形成氢键,苯胺环与FAK的Leu567有疏水作用,这些作用都可稳定DFG超二级结构的螺旋构象;TAE226与FAK结合后可引起Asp564相对于在FAK活性状态时的位置发生113°φ扭转角,可见TAE226对FAK有很好的选择性抑制作用[30]。但其不是靶向FAK的特效药,且目前尚无有关其临床试验的报道。
3.1.4 PND-1186将TAE226嘧啶环用吡啶环取代,其原C5位的氯用三氟甲基取代,即得另一FAK抑制剂PND-1186(4)。本品可选择性抑制FAK活性,促进癌细胞凋亡,对FAK的体外IC50为1.5nmol·L-1,对乳腺癌细胞的IC50约为100nmol·L-1[2]。本品不影响黏着细胞中c-Src和p130Cas酪氨酸的磷酸化,但对非黏着状态的细胞,其可阻抑FAK和130Cas酪氨酸的磷酸化,提高细胞凋亡蛋白酶的活性,导致细胞凋亡[31]。

3.2 ATP非依赖型FAK小分子抑制剂
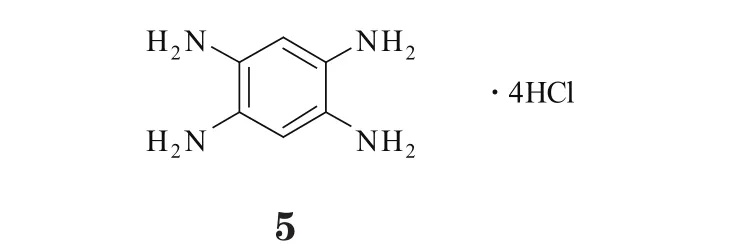
Y15(5)即为ATP非依赖型FAK小分子抑制剂,其发挥对FAK的抑制作用时,并不通过ATP结合位点,而是靶向抑制FAK的Y397位自磷酸化。体外实验表明,Y15可剂量和时间依赖性地减少FAK的表达和激活。小鼠异种移植瘤模型实验显示,Y15可有效抑制细胞流动性及肿瘤的形成和生长,促进细胞分离和凋亡,且耐受性良好,经口和腹膜注射给药均无明显毒性[32]。体内实验表明,Y15可显著减小注射结肠癌SW620细胞所形成的肿瘤模型小鼠的肿瘤体积,其治疗结肠癌的疗效甚至好于标准化疗药物5-氟尿嘧啶,且它能提高癌细胞对5-氟尿嘧啶的敏感性,与化疗药物合用时可产生协同作用[33]。此外,Y15对许多类型的肿瘤(如神经细胞瘤、乳腺癌、胰腺癌和转移性的结肠癌等)均能产生治疗作用。有研究者通过对Y15进行结构改造,获得了能更好靶向FAK自磷酸化位点的若干衍生物。其中,Y15通过FAK上自磷酸化位点的上游靶点来产生对FAK激酶的特异性抑制作用,同时还可阻抑Src发生自身磷酸化而抑制其功能。体外实验显示,Y15能通过抑制FAK而显著降低胶质母细胞瘤细胞生长和生存能力。在神经胶质瘤模型小鼠中进行的体内实验显示,与对照组相比,经Y15治疗的模型小鼠其肿瘤明显缩小,生存期也大大延长,尤其是Y15与标准化疗药物替莫唑胺联用可产生协同作用,疗效更佳[34]。
Y15为四苯胺盐酸盐化合物,其苯环上的碳原子可与FAK的Gly59产生范德华力相互作用,而3个氨基可分别与FAK的Arg57、Glu399和Thr394形成氢键。研究表明,Y15可显著降低全长FAK的催化区域活性,对其他激酶的活性影响较小;Y15对缺少包括Y397位点的N端区域的FAK激酶活性几乎无影响,同样它对FAK同源蛋白Pyk2的活性和ATP结合位点也无影响[32]。可见,Y15是直接的特异性FAKY397位自磷酸化抑制剂。
此外,还有其他已完成临床Ⅰ期试验的FAK小分子抑制剂,如PF04554878和GSK2256098,不过它们的结构目前均未见报道。临床试验发现,GSK2256098可抑制Ⅱ型神经纤维瘤(NF2)基因失活的间皮细胞瘤病人肿瘤扩散[35],而PF04554878能有效治疗早期的非血液系统恶性肿瘤[36]。在我国,也有学者专注于FAK抑制剂的研究,并发现多种具有FAK抑制活性的母核结构,例如吡咯并[2,3-d]嘧啶[37]、1,3,4-噁二唑[38]和7-巯基黄酮等等。
4 结语
FAK是一种非受体型酪氨酸蛋白激酶,在许多肿瘤的发生和发展过程中均有过表达,其作为抗肿瘤治疗的有效靶点备受关注。自2005年开始将FAK作为抗肿瘤靶点研究以来,科学家揭示了该靶点的一些普遍特征,并通过对FAK抑制剂与FAK的复合物晶体结构的分析,发现作用于FAK的Y397自磷酸化位点可有效提高FAK抑制剂的选择性,这为深入研究新型高效、高选择性的FAK小分子抑制剂提供了极具价值的理论指导。而且,多个颇具治疗前景的FAK小分子抑制剂也陆续开发进入临床前和临床研究,但目前尚无此类药物上市。
[1]Lechertier T, Hodivala-Dilke K. Focal adhesion kinase and tumour angiogenesis[J]. J Pathol, 2012, 226(2): 404-412.
[2]Mitra S K, Schlaepfer D D. Integrin-regulated FAK-Src signaling in normal and cancer cells[J]. Curr Opin Cell Biol, 2006, 18(5): 516-523.
[3]Chen S Y, Chen H C. Direct interaction of focal adhesion kinase (FAK) with Met is required for FAK to promote hepatocyte growth factorinduced cell invasion[J]. Mol Cell Biol, 2006, 26(13): 5155-5167.
[4]Lim S T, Mikolon D, Stupack D G, et al. FERM control of FAK function: implications for cancer therapy[J]. Cell Cycle, 2008, 7(15): 2306-2314.
[5]Chatzizacharias N A, Kouraklis G P,Theocharis S E. Focal adhesion kinase: a promising target for anticancer therapy[J]. Expert Opin Ther Targets, 2007, 11(10): 1315-1328.
[6]Halder J, Lin Y G, Merritt W M, et al.Therapeuticeffcacyofanovelfocal adhesion kinase inhibitor TAE226 in ovarian carcinoma[J]. Cancer Res, 2007, 67(22): 10976-10983.
[7]McLean G W, Carragher N O, Avizienyte E, et al. The role of focaladhesion kinase in cancer - a new therapeutic opportunity[J]. Nat Rev Cancer, 2005, 5(7): 505-515.
[8]Mitra S K, Hanson D A,Schlaepfer D D. Focal adhesion kinase: in command and control of cell motility[J]. Nat Rev Mol Cell Biol, 2005, 6(1): 56-68.
[9]Chen J S, Huang X H, Wang Q, et al. Sonic hedgehog signaling pathway induces cell migration and invasion through focal adhesion kinase/AKT signaling-mediated activation of matrix metalloproteinase (MMP)-2 and MMP-9 in liver cancer[J]. Carcinogenesis, 2013, 34(1): 10-19.
[10]Chen J S, Huang X H, Wang Q, et al. FAK is involved in invasion and metastasis of hepatocellular carcinoma[J]. Clin Exp Metastasis, 2010, 27(2): 71-82.
[11]Lipinski C A, Tran N L, Bay C, et al. Differential role of proline-rich tyrosine kinase 2 and focal adhesion kinase in determining glioblastoma migration and proliferation[J]. Mol Cancer Res, 2003, 1(5): 323-332.
[12]Liu W, Bloom D A, Cance W G, et al. FAK and IGF-IR interact to provide survival signals in human pancreatic adenocarcinoma cells[J]. Carcinogenesis, 2008, 29(6): 1096-1107.
[13]Canel M, Secades P, Rodrigo J P, et al. Overexpression of focal adhesion kinase in head and neck squamous cell carcinoma is independent of fak gene copy number[J]. Clin Cancer Res, 2006, 12(11 Pt 1): 3272-3279.
[14]Chan K T, Cortesio C L,Huttenlocher A. FAK alters invadopodia and focal adhesion composition and dynamics to regulate breast cancer invasion[J]. J Cell Biol, 2009, 185(2): 357-370.
[15]Xia J, Lv N, Hong Y, et al. Increased expression of focal adhesion kinase correlates with cellular proliferation and apoptosis during 4-nitroquinoline-1-oxide-induced rat tongue carcinogenesis[J]. J Oral Pathol Med, 2009, 38(6): 524-529.
[16]Canel M, Secades P, Garzon-Arango M, et al. Involvement of focal adhesion kinase in cellular invasion of head and neck squamous cell carcinomas via regulation of MMP-2 expression[J]. Br J Cancer, 2008, 98(7): 1274-1284.
[17]Sood A K, Armaiz-Pena G N, Halder J, et al. Adrenergic modulation of focal adhesion kinase protects human ovarian cancer cells from anoikis[J]. J Clin Invest, 2010, 120(5): 1515-1523.
[18]Kohno M, Hasegawa H, Miyake M, et al. CD151 enhances cell motility and metastasis of cancer cells in the presence of focal adhesion kinase[J]. Int J Cancer, 2002, 97(3): 336-343.
[19]Halder J, Landen C N Jr, Lutgendorf S K, et al. Focal adhesion kinase silencing augments docetaxel-mediated apoptosis in ovarian cancer cells[J]. Clin Cancer Res, 2005, 11(24 Pt 1): 8829-8836.
[20]Halder J, Kamat A A, Landen C N Jr, et al. Focal adhesion kinase targeting using in vivo short interfering RNA delivery in neutral liposomes for ovarian carcinoma therapy[J]. Clin Cancer Res, 2006, 12(16): 4916-4924.
[21]Li S, Dong W, Zong Y, et al. Polyethylenimine-complexed plasmid particles targeting focal adhesion kinase function as melanoma tumor therapeutics[J]. Mol Ther, 2007, 15(3): 515-523.
[22]Earley S, Plopper G E. Disruption of focal adhesion kinase slows transendothelial migration of AU-565 breast cancer cells[J]. Biochem Biophys Res Commun, 2006, 350(2): 405-412.
[23]Roberts W G, Ung E, Whalen P, et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,27[J]. Cancer Res, 2008, 68(6): 1935-1944.
[24]Stokes J B, Adair S J, Slack-Davis J K, et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment[J]. Mol Cancer Ther, 2011, 10(11): 2135-2145.
[25]Slack-Davis J K, Martin K H, Tilghman R W, et al. Cellular characterization of a novel focal adhesion kinase inhibitor[J]. J Biol Chem, 2007, 282(20): 14845-14852.
[26]Bagi C M, Christensen J, Cohen D P, et al. Sunitinib and PF-562,271 (FAK/Pyk2 inhibitor) effectively block growth and recovery of human hepatocellular carcinoma in a rat xenograft model[J]. Cancer Biol Ther, 2009, 8(9): 856-865.
[27]Dhani N C, Burris H A III, Siu L L, et al. Final report of phase I clinical, pharmacokinetic (PK), pharmacodynamic (PD) study of PF-00562271 targeting focal adhesion kinase (FAK) in patients (pts) with solid tumors[J]. J Clin Oncol, 2010, 28(15 suppl): abstr 3028.
[28]Jones M L, Shawe-Taylor A J, Williams C M, et al. Characterization of a novel focal adhesion kinase inhibitor in human platelets[J]. Biochem Biophys Res Commun, 2009, 389(1): 198-203.
[29]Wang Z G, Fukazawa T, Nishikawa T, et al. TAE226, a dual inhibitor for FAK and IGF-IR, has inhibitory effects on mTOR signaling in esophageal cancer cells[J]. Oncol Rep, 2008, 20(6): 1473-1477.
[30]Lietha D, Eck M J. Crystal structures of the FAK kinase in complex with TAE226 and related bis-anilino pyrimidine inhibitors reveal a helical DFG conformation[J]. PLoS One, 2008, 3(11): e3800.
[31]Walsh C, Tanjoni I, Uryu S, et al. Oral delivery of PND-1186 FAK inhibitor decreases tumor growth and spontaneous breast to lung metastasis in pre-clinical models[J]. Cancer Biol Ther, 2010, 9(10): 778-790.
[32]Golubovskaya V M, Nyberg C, Zheng M, et al. A small molecule inhibitor, 1,2,4,5-benzenetetraamine tetrahydrochloride, targeting the y397 site of focal adhesion kinase decreases tumor growth[J]. J Med Chem, 2008, 51(23): 7405-7416.
[33]Wang S, Basson M D. Akt directly regulates focal adhesion kinase through association and serine phosphorylation: implication for pressure-induced colon cancer metastasis[J]. Am J Physiol Cell Physiol, 2011, 300(3): C657-670.
[34]Golubovskaya V M, Huang G, Ho B, et al. Pharmacologic blockade of FAK autophosphorylation decreases human glioblastoma tumor growth and synergizes with temozolomide[J]. Mol Cancer Ther, 2013, 12(2): 162-172.
[35]Soria J-C, Gan H K, Arkenau H-T, et al. Phase I clinical and pharmacologic study of the focal adhesion kinase (FAK) inhibitor GSK2256098 in pts with advanced solid tumors[J]. J Clin Oncol, 2012, 30(15 suppl): abstr 3000.
[36]Jones S, Shapiro G, Bendell J, et al. Phase I study of PF-04554878, a second-generation focal adhesion kinase (FAK) inhibitor, in patients with advanced solid tumors[J]. J Clin Oncol, 2011, 29(15 suppl): abstr 3002.
[37]Choi H S, Wang Z, Richmond W, et al. Design and synthesis of 7H-pyrrolo[2,3-d]pyrimidines as focal adhesion kinase inhibitors. Part 1[J]. Bioorg Med Chem Lett, 2006, 16(8): 2173-2176.
[38]Zhang S, Luo Y, He L Q, et al. Synthesis, biological evaluation, and molecular docking studies of novel 1,3,4-oxadiazole derivatives possessing benzotriazole moiety as FAK inhibitors with anticancer activity[J]. Bioorg Med Chem, 2013, 21(13): 3723-3729.
Research Progress on Focal Adhesion Kinase and Its Small Molecule Inhibitors as Antitumor Agents
YANG Chao, WANG Chongqing, CHEN Ying, TIAN Wei, ZHU Ju
( Department of Medicinal Chemistry, School of Pharmacy, NO.2 Military Medical University, Shanghai 200433, China)
Focal adhesion kinase (FAK) is a non-receptor tyrosine kinase overexpressed in the development and progression of many tumors. It has been suggested that as an important intracellular cytoskeletal protein and a key molecule in a variety of cell signal transduction pathways, FAK playsasignifcantroleintumorcellsurvival,development,migrationandinvasion.Therefore,theresearchesonthedevelopmentofFAKinhibitorsas antitumor agents have attracted an extensive attention. The structure and function of FAK, its association with tumor and research and development of its small molecule inhibitors were reviewed.
focal adhesion kinase; cell signal transduction pathway; target; small molecular FAK inhibitor; antitumor activity
R34; R979.1
A
1001-5094(2014)09-0649-07
接受日期:2014-07-29
*通讯作者:朱驹,教授,博士生导师;
研究方向:药物设计与合成;
Tel:021-81871238;E-mail:zhuju@smmu.edu.cn
猜你喜欢
波谱学杂志(2022年1期)2022-03-15
天津医科大学学报(2021年3期)2021-07-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
天津医科大学学报(2019年6期)2019-08-13
天然产物研究与开发(2018年7期)2018-08-21
分析化学(2017年12期)2017-12-25
上海农业学报(2017年3期)2017-04-10
中国医药生物技术(2015年4期)2015-12-26
安徽医科大学学报(2015年9期)2015-12-16
中国当代医药(2015年16期)2015-03-01
