
新型多靶向他克林衍生物的研究进展
2014-03-08 02:07陈瑶孙昊鹏李伟
药学进展 2014年9期
陈瑶,孙昊鹏,李伟*
(1. 南京中医药大学药学院,江苏 南京 210023; 2. 中国药科大学药学院,江苏 南京 210009)
新型多靶向他克林衍生物的研究进展
陈瑶1,孙昊鹏2,李伟1*
(1. 南京中医药大学药学院,江苏 南京 210023; 2. 中国药科大学药学院,江苏 南京 210009)
阿尔茨海默病(AD)是一种常见的神经退行性疾病,不仅严重威胁着老年人的身心健康,更给整个社会医疗卫生系统带来沉重负担,因此,设计与开发安全有效的抗AD药物,一直是医药研究领域的热点。AD病因尚不明确且致病机制极为复杂,仅针对单一靶标治疗难以获得理想疗效,因此一药多靶成为了抗AD药物研发的新方向。目前临床上最为成功的抗AD药物为乙酰胆碱酯酶 (AChE)抑制剂,尽管第一代AChE抑制剂他克林因严重的毒副作用而撤市,但其结构简单,活性明确,在用于多靶向设计的结构改造方面具有独特优势,可作为理想的活性片段,引入多靶向抗AD药物的整体分子骨架中。通过一些代表性案例,综述近年来基于他克林结构而设计多靶向抗AD药物的研究进展,探讨其设计思路,为新型多靶向他克林衍生物的研发提供参考。
阿尔茨海默病;乙酰胆碱酯酶抑制剂;他克林;多靶向衍生物;分子设计
阿尔茨海默病(Alzheimer’sdisease,AD)于1906年由德国医生AloisAlzheimer发现并报道,而今,它已经成为老龄化社会的标志性疾病之一,严重威胁着老年人的身心健康。根据国际阿尔茨海默症协会的报道,目前全球AD患者人数超过3600万,预计到2050年这一数字会增加到1.15亿,其中绝大多数患者分布在中低收入国家[1]。AD不仅给患者及家人带来病痛和精神的折磨,更给社会医疗卫生系统造成沉重负担,据统计,2010年AD导致的直接和间接花费高达6040亿美元,接近世界GDP的1%[2-3]。我国的AD防治形势同样十分严峻,目前我国AD患者约有600~700万人,20个老年人中就有一个AD患者,约占世界总病例的1/4,我国已成为世界AD患者的第一大国。可见,开发安全有效的AD治疗药物,无论对于中国还是全世界,都已经刻不容缓。
AD的病理成因极为复杂,至今仍未发现其明确的致病因素,其病理进程中更是涉及到众多靶标与信号通路。长久以来,医学界对于AD的致病模型存有很大争议,这也直接阻碍了抗AD药物的研发。尽管目前科学家对AD确切病因尚无定论,但前期大量研究已表明,AD的病理演变涉及神经、免疫以及血液循环等多个系统和环节,其诱因包括:1)脑部微循环萎缩和供血不足;2)引起脑部体液内环境变化的炎症反应及脑部活泼氧自由基水平的上升;3)直接导致AD症状的脑部胆碱神经递质生成不足、功能紊乱以及胆碱受体敏感度下降,即胆碱能假说(cholinergichypothesis);4)诱导神经元细胞凋亡的β-淀粉样蛋白(β-amyloid,Aβ)缠结、沉淀;5)引起神经纤维缠结的Tau蛋白过度磷酸化,致使脑内神经元丧失和神经递质水平降低、老年斑(senileplaque,SP)形成以及神经元纤维缠结(neurofibrillarytangles,NFT),最终导致脑部神经系统的整体性病变[4]。
目前的抗AD药物研发处于相当窘迫的境地。一方面,临床上可供选择的抗AD药物品种非常有限,自2003年最后一个抗AD药物被美国食品药品管理局(FDA)批准以来,总共只有6个上市药物,分别为乙酰胆碱酯酶(AChE)抑制剂他克林(tacrine,1)、多奈哌齐(donepezil,2)、利凡斯的明(rivastigmine,3)、石杉碱甲[(-)-huprineA,4]和加兰他敏(galanthamine,5)以及N-甲基-D-天冬氨酸(NMDA)受体阻滞剂美金刚(memantine,6),且这些药物用于治疗AD时仅能延缓发病进程,并无根治作用,尤其对中晚期AD病人的疗效不佳,只适用于轻、中度AD的对症治疗[5]。


另一方面,针对AD发病过程中各种靶点的100多个候选药物相继开发失败。例如,由礼来(Lilly)公司研发的γ-分泌酶抑制剂semagacestat于2010年被终止开发,原因是,其对血脑屏障(bloodbrainbarrier,BBB)的透过率低,且难以避免对Notch通路的影响,从而产生毒副作用[6];曾被寄予厚望用作Aβ清除剂的单克隆抗体(单抗)药物——辉瑞公司(Pfizer)与强生公司(Johnson&Johnson)联合开发的bapineuzumab和礼来公司开发的solanezumab,由于无明确的AD治疗效果,于2013年被相继宣布终止于Ⅲ期临床研究阶段[7],而更值得注意的是,这些单抗药物虽然可几乎完全清除AD患者脑部的Aβ,却依然无法逆转AD,这进一步说明了AD致病因素的复杂性。这些失败的研发案例虽有负面效应,但也促使人们重新审视AD的治疗策略,显然,面对如此复杂的系统性疾病,只针对某一个靶点或环节的单靶向药物难以实现对整体疾病网络的持续性调节,任何的内源性代偿调节方式都有可能造成单靶向药物丧失靶标而失活,这也解释了已上市的抗AD药物无法获得理想疗效的原因。因此,近年来,针对AD发病进程中的多重靶标,设计多功能抑制剂(即一药多靶),已成为抗AD药物研发的全新方向。
辉瑞公司开发的他克林于1993年被美国FDA批准上市,成为第一个临床有效的抗AD药物,虽然后来由于会导致严重的肝毒性而被迫退出市场,但笔者认为,不能忽视其在多靶向抗AD药物设计中的价值。首先,相较于其他上市药物如多奈哌齐,他克林结构简单,相对分子质量仅为198.27,具有理想的类药性和药代动力学特性,且可耐受大幅度的结构改造而保持对AChE的抑制活性,从而为其提供了广阔的结构改造空间;其次,他克林合成工艺简单、可控,相较于来源稀缺、合成困难的天然产物如石杉碱甲,在工业生产及成本控制方面具有明显的优势;第三,他克林活性非常确切,对AChE具有强效抑制作用(IC50为10~50nmol·L-1)[8-10],分子的配体效率(ligandefficiency,LE)很高,是理想的活性片段结构;最后,他克林引发的肝毒性与其游离的伯氨基密切相关,而在药物多靶向设计中,该氨基可通过其他功能片段的引入而被修饰,从而大幅度降低分子的肝毒性,因此他克林引发的肝毒性是一个可在药物多靶向设计中被规避的问题。
多靶向抗AD药物由于需要同时针对数个AD致病环节,其分子结构中势必包含多个功能片段,导致分子复杂性提高及相对分子质量增大,其类药性降低。因此,如何选择功能片段以及如何对其加以合理的组合和调配,对于药物多靶向的设计策略至关重要。笔者认为,活性明确、结构简单的他克林非常适合作为AChE抑制的功能单元而融入多靶向药物的整体分子骨架中,其在多靶向抗AD药物的研发中具有独特优势。本文通过一些代表性案例,并结合笔者自身的研究工作,综述近年来基于他克林而设计多靶向抗AD药物的研究进展,探讨其设计思路,为新型多靶向他克林衍生物的研发提供参考。
1 乙酰胆碱酯酶的结构生物学与其靶向双联他克林衍生物的设计
AChE广泛分布于人脑中各种神经肌肉组织,如黑质、海马、尾状核、小脑等,其生理上最主要的功能是,迅速降解神经递质乙酰胆碱(ACh),进而维持突触间隙ACh浓度的平衡。1991年,第1个电鳐源的AChE晶体结构(ProteinDataBank,PDB,ID:2ACK)被解析出来(Sussman等,Science,1991年)。随后,鼠源、电鳗源和人源的AChE晶体结构也相继被报道(Bourne等,J Biol Chem,1999年;Kryger等,Acta Crystallogr D Biol Crystallogr,2000年;Bourne等,EMBO J,2003年)。目前,PDB数据库已收录了各类AChE结构168个,它们为深入了解AChE的结构特征、进而开展相关药物分子设计奠定了坚实的基础。空间上,AChE呈现近球状三维结构(见图1A),其配体结合域(ligandbindingdomain,LBD)由蛋白底部的表面亲水区一直延伸至内部疏水腔,跨距约为2nm。LBD主要可分为两部分,其一是位于疏水腔内部的活性催化位点(catalyticsite,CAS),包含由Ser200、Glu327和His440所组成的催化三联体,主要完成对ACh的水解,已上市的AChE抑制剂均可稳定结合于这一位点,从而阻断AChE的催化功能;其二是位于蛋白浅表开口处的外周阴离子位点(peripheralanionicsite,PAS),主要包含Tyr70、Trp279等芳香性残基,它们可与阳离子形成阳离子-π相互作用,也可与环状结构形成是π-π堆叠作用,从而稳定配体与AChE的结合(见图1B)。点突变实验表明,PAS突变可导致AChE抑制剂活性显著下降。此外,PAS被证实与AChE诱导的Aβ聚集有关。因此,阻断PAS,有助于减少神经纤维缠结的形成。
分析AChE与他克林的结合模式(见图1C)可以看出,他克林与CAS中的Trp84和Phe330之间通过π-π堆叠作用形成较为稳定的“三明治”式构象,伯氨基与周围临近的水分子形成氢键网络,促进了结合的稳定性,而母环上的N原子与His440靠近,可干扰AChE对ACh的水解。此结合模式从原子层面上清楚地解释了他克林对AChE具有高效抑制活性的原因。
2006年,Rydberg等[11]报道了一类全新结构的双联他克林衍生物(7),即以烷基二胺侧链为连接臂,将两分子的他克林加以连接。晶体结构分析表明,双联他克林以合理的构象嵌入AChE的LBD之中,其中一分子他克林与CAS结合,而另一分子则与PAS中的Tyr70、Trp279形成“三明治”式π-π堆叠作用(见图1D)。该化合物与AChE的双位点(dualsites)结合模式大大增强了结合稳定性,因此其活性相比于他克林提高了约1400倍,且肝毒性显著降低。
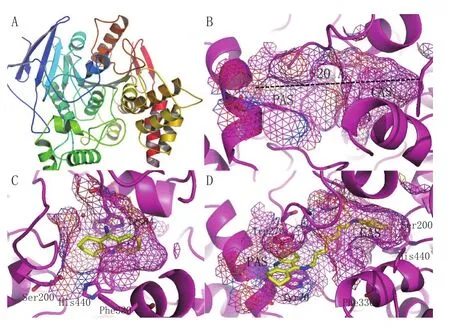
图 1 AChE及其与药物结合的复合物晶体结构Figure 1 Crystal structures of AChE and its complexes with drugs
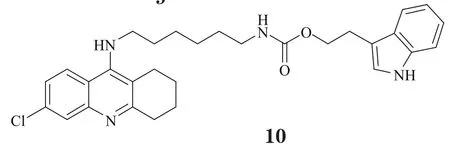
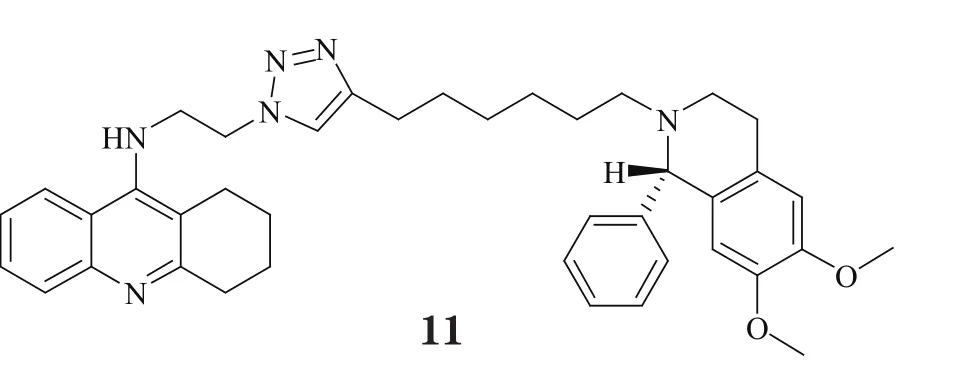
以上研究结果充分说明了CAS与PAS在AChE的LBD中所起作用,同时也提示,适宜的疏水结构能与PAS上芳香性残基形成良好的结合,而且若其能通过合适的连接臂与CAS结合片段相连,即可达到双位点抑制效果。受此思路的启发,学界掀起了对双位点型AChE抑制剂研发的热潮,尤其是对新型双联他克林衍生物的发掘。Alonso等[12]把他克林分子与多奈哌齐的苄基哌啶部分相连,Shao等[13]则将他克林与多奈哌齐的茚酮相连,并对其连接臂以及片段结构进行局部改造,得到了两类活性优于他克林的全新骨架(8,9),且它们对AChE具有更好的选择性。Martinez研究团队将他克林母环和吲哚进行分子融合,并在连接臂中引入酰胺结构(10),从而进一步增强了化合物对AChE的识别能力,生物活性可达皮摩尔级[14]。此外,Krasinski等[15]还在双联他克林衍生物的连接臂中引入杂环,合成了一系列具有立体选择性的AChE抑制剂,其中化合物11对AChE的抑制活性最好,甚至达到飞摩尔水平,是目前报道的体外活性最强的非共价结合型AChE抑制剂。
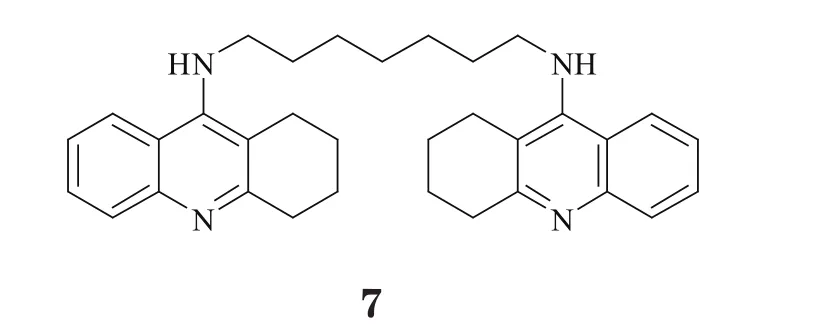

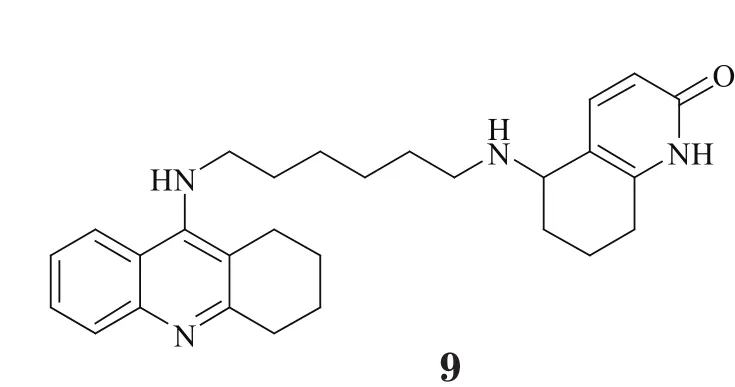
2 多靶向他克林衍生物的设计
近年来,通过多靶向策略治疗AD,已逐渐成为共识。上述双联他克林衍生物的设计在分子水平上为多靶向他克林衍生物的设计奠定了基础,即以他克林为AChE结合片段,引入不同类型的功能片段,调节其他重要的AD治疗靶标活性,从而获得多靶向抗AD药物。与他克林相比,这些多靶向衍生物具有更强的抗AD活性,同时大大降低了肝毒性。
2.1 乙酰胆碱酯酶-分泌酶双靶向他克林衍生物大量研究表明,Aβ在AD的发展进程中扮演重要角色,它是老年斑的主要成分,易于聚集而形成球状的寡聚体、纤维、斑块,进而产生强烈的神经毒性,造成神经细胞损伤乃至死亡,最终造成AD病人严重的认知障碍。Aβ具有多种聚合方式及类型,通常认为其低聚物具有更强的神经毒性,这是由于它能造成Tau蛋白过度磷酸化,加剧神经纤维缠结的形成。因此,阻止Aβ低聚物的形成,抑制其聚合能力,成为了抗AD药物靶向研究的热点。而Aβ的生成和聚合主要在分泌酶家族蛋白的催化下完成,故药物研究人员设计了多种类型的AChE-分泌酶双靶向他克林衍生物。
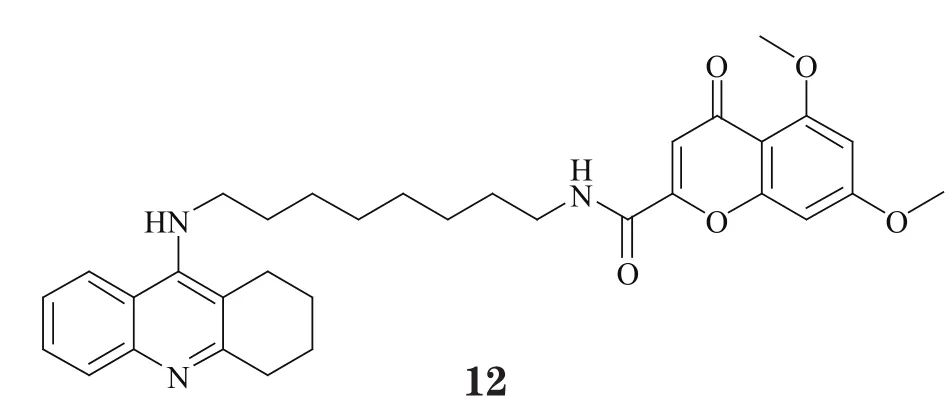
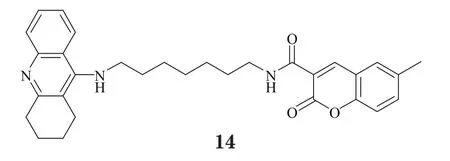
Fernández-Bachiller等[16]将他克林通过烷基二胺侧链与对β-分泌酶(BACE)具有抑制作用的色酮环相连,得到了一系列化合物,其中代表性化合物12对AChE和BACE均具有良好的抑制作用,对它们的IC50分别为2.3nmol·L-1和2.1μmol·L-1。Rizzo等[17]对他克林的骨架结构进行了改造,将其中的环己烷替换为2,3-二氢茚环,并将两分子改造骨架以烷基二胺侧链相连接,获得一类新型双靶向抑制剂(13),其虽对AChE的抑制活性一般(IC50=455.0nmol·L-1),却具有较好的BACE抑制活性(IC50=0.4μmol·L-1)。Sun等[18]将香豆素片段与他克林连接,得到具有中等双靶向抑制活性的化合物(14),其对AChE的Ki为66.1nmol·L-1,对BACE的IC50为42.8μmol·L-1,且值得一提的是,该化合物对Aβ生成具有明显的抑制作用(IC50=6.1μmol·L-1),强于香豆素(IC50=11.0μmol·L-1),这也说明多靶向药物在协同治疗方面的优势。
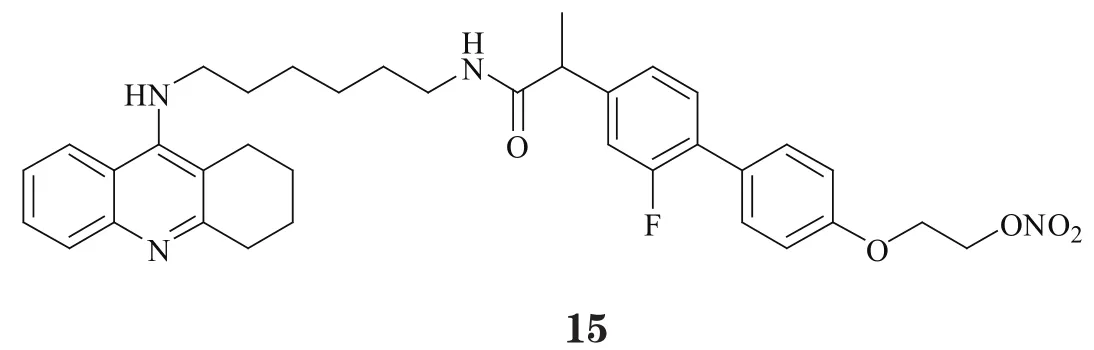
在Aβ生成过程中,另一个关键靶标是γ-分泌酶。笔者将对γ-分泌酶具有明确抑制活性的非甾体抗炎药氟比洛芬作为结构片段,与他克林通过烷基二胺相连,得到一类具有AChE-γ-分泌酶双靶向抑制活性的化合物,其中化合物15对AChE的IC50为9.1nmol·L-1,且在0.5μmol·L-1的浓度下对Aβ聚集的抑制率为17.0%[19],虽然活性稍弱,但为多靶向抗AD药物的设计提供了一种全新的思路。

2.2 乙酰胆碱酯酶-抗氧化双靶向他克林衍生物
随着对AD病理机制研究的不断深入,人们意识到,除胆碱能损伤和Aβ异常聚集外,AD病人脑部异常生成的超氧自由基同样会损伤脑部神经元。虽然这些超氧自由基究竟由何种原因诱导生成,目前尚无定论,但其引起的氧化应激(oxidativestress)对神经元的损伤是非常明确的,氧化应激能促进Tau蛋白的异常磷酸化,加速神经纤维缠结,进而促使AD的形成。因此,开发具有抗氧化作用的多靶向抗AD药物,也是一种有价值的治疗策略。
8-羟基喹啉具有良好的金属离子螯合能力和还原性,是常见的药物优势骨架。Fernández-Bachiller[20]将他克林与8-羟基喹啉通过烷基二胺侧链相连,得到化合物16,其不仅能强烈抑制AChE(IC50=1.0nmol·L-1),同时对超氧自由基具有良好的吸收及清除能力,氧自由基吸收能力(oxygenradicalabsorbancecapacity, ORAC)测试表明,其对氧自由基的相对吸收值可达4.7。
阿魏酸在体内具有明确的抗氧化活性,可强效清除氧化自由基,有效保护细胞免受氧化损伤。Fang 等[21]进行的实验研究显示,小鼠长期口服阿魏酸,能有效降低Aβ42 的生成及其造成的神经细胞毒性,对小鼠智力具有一定的改善作用。于是,该研究小组以烷基二胺侧链将他克林与阿魏酸相连,结果发现,所得偶联物(17)在保持AChE 抑制活性的同时,还具有一定的抗氧化能力,ORAC 测试的相对吸收值为2.0。
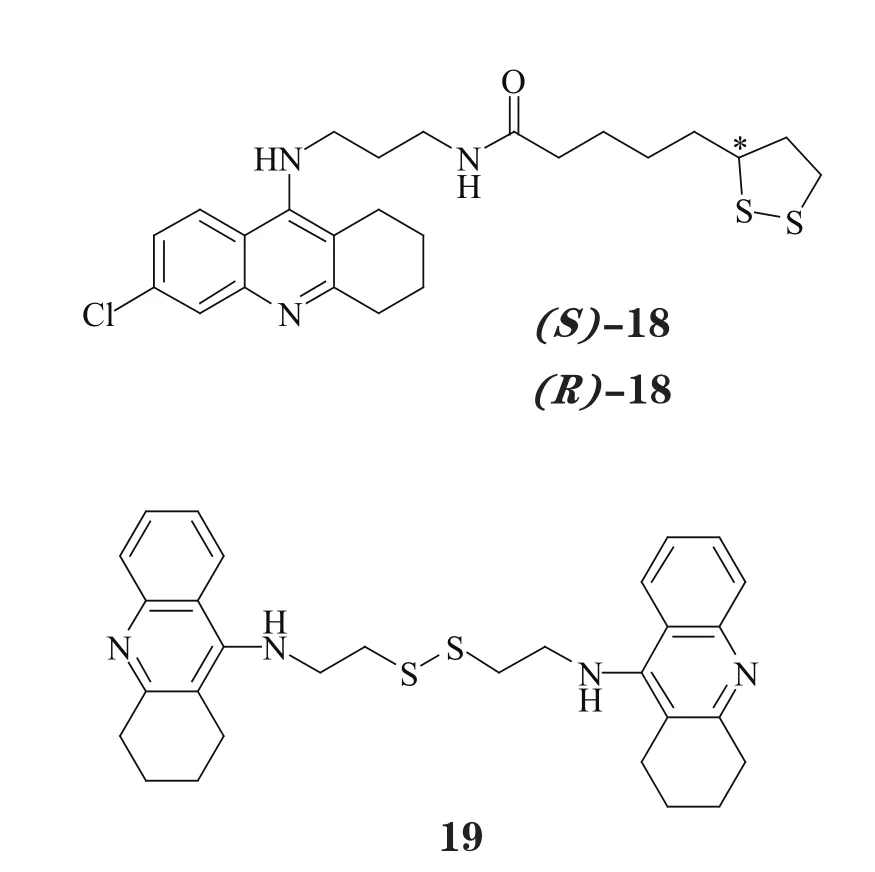
硫辛酸(lipoic acid,LA)是具有抗氧化活性的天然产物,其结构中的二硫键具有很强的还原能力,可实现对氧自由基的清除。Rosini 等[22]设计了他克林与LA的偶联物,并进一步将其拆分,得到了光学纯化合物(18),研究表明,其对映体具有非常接近的强AChE抑制活性,(S)-18 和(R)-18 对AChE 的IC50分别为 0.23和0.47 nmol·L-1,且其在0.5 μmol·L-1的低浓度下即能对抗H2O2引起的细胞损伤和凋亡。目前,化合物18已成为AD 病理研究中的首选工具药之一,其多项体内研究已开展,是非常有前景的抗AD 候选药物之一。化合物19 也是以相似策略设计的双联他克林衍生物,对AChE 和Aβ 聚集的IC50分别为5.0 nmol·L-1和24.2 μmol·L-1,并同样在0.5 μmol·L-1的低浓度下即能逆转H2O2的氧化应激损伤[23]。
咔唑因具有明确的抗氧化能力,近年来也引起了药物化学家的广泛关注。Thiratmatrakul 等[24]设计了一类结构独特的他克林- 咔唑偶联衍生物(20),其对AChE 的抑制活性虽一般(IC50= 0.95 μmol·L-1),却具有较强的抗氧化能力,2,2′- 联氮- 双(3- 乙基苯并噻唑啉-6- 磺酸)(ABTS)法测试显示其清除氧自由基的EC50 为11.2 μmol·L-1,并能显著增强细胞在H2O2环境中的生存能力。



2.3 一氧化氮供体型多靶向他克林衍生物
一氧化氮(NO)是体内重要的信使分子,具有广泛的生理作用。近来研究表明,NO在AD的发病进程中同样起关键作用。AD病人往往伴随着脑部微循环障碍,并引起内源性NO水平下降,而NO不足又进一步导致脑部血流量下降,造成微循环系统持续失控,但若能适量补充外源性NO,可有效恢复神经系统中NO平衡,阻断AD的病理进程。
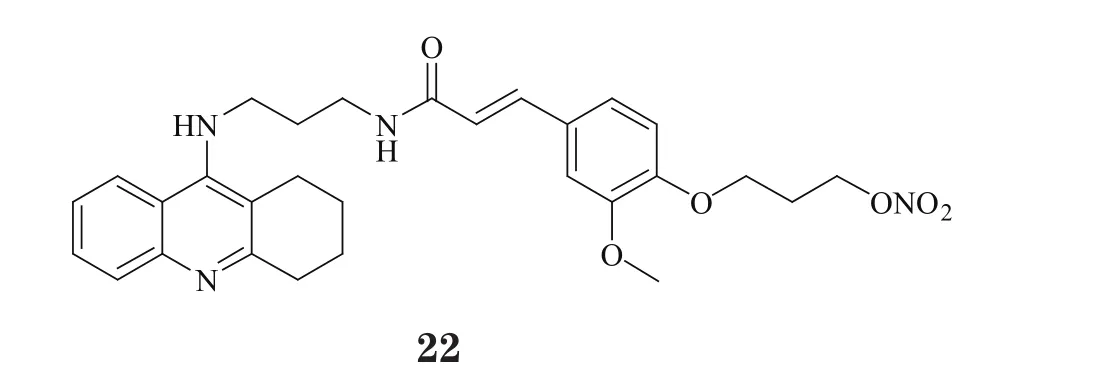
笔者所在研究团队对NO供体药物进行了多年研究。在此基础上,笔者通过引入硝酸酯基团作为NO供体,设计、合成了一系列NO供体型多靶向他克林衍生物。体外试验表明,其中代表性化合物21和22不仅能高效抑制AChE活性(IC50分别为5.6和10.9nmol·L-1),还具有明显的血管舒张作用,活性与阳性对照药物二硝酸异山梨酯(isosorbidedinitrate,ISDN)相当。体内实验表明,这两个化合物可显著改善东莨菪碱所致大鼠认知功能障碍。值得关注的是,相比于他克林,化合物22的肝毒性显著降低,对肝脏天冬氨酸氨基转移酶、血浆蛋白及总蛋白的含量几乎无影响,提示该化合物具有良好的安全性[25-26]。

2.4 其他类型的多靶向他克林衍生物
近年来,随着对AD病理机制研究的不断深入,人们发现了一些新的AD治疗靶标,为多靶向抗AD药物的研发提供了理论依据,且针对这些新靶标而设计的多靶向他克林衍生物也相继被报道。
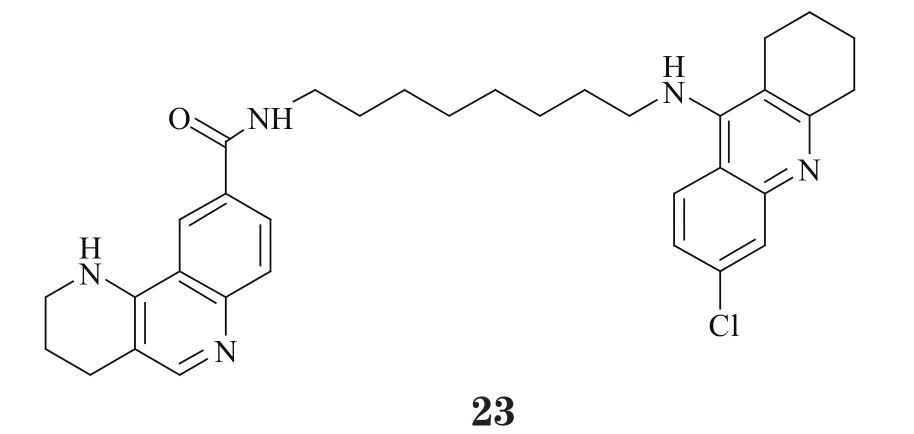
2.4.1 对Tau蛋白聚集具有抑制作用的多靶向他克林衍生物DiPietro等[27]报道了哌啶并萘啶骨架与他克林偶联形成的化合物(23),其显著特点是,在抑制AChE的同时(IC50=2.0nmol·L-1),还能阻止Tau蛋白的聚集(10μmol·L-1浓度下的抑制率达68.7%),并可显著抑制Aβ42的聚集(10μmol·L-1浓度下的抑制率达77.5%)。提示,这是一种思路较为新颖的多靶向设计策略。
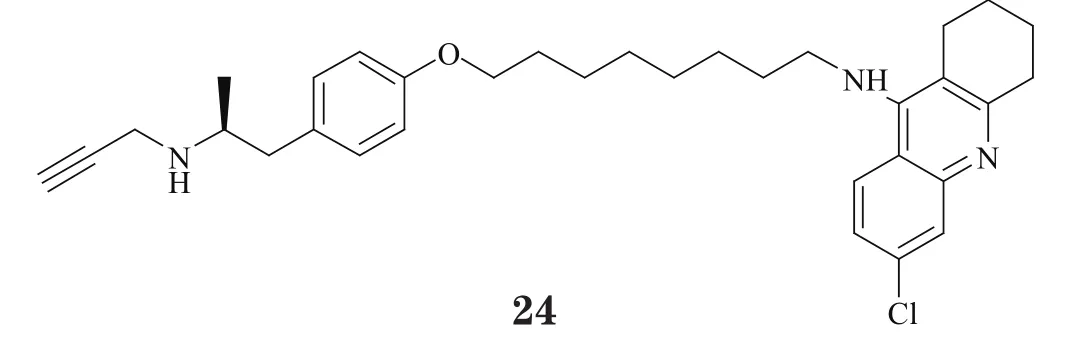
2.4.2 对单胺氧化酶具有抑制作用的多靶向他克林衍生物单胺氧化酶(MAO)是人脑内重要的神经递质代谢酶,主要分布在神经元和星形胶质细胞中,负责对单胺类神经递质(如儿茶酚胺、5-羟色胺等)的氧化,从而调控其动态平衡。临床研究表明MAO与包括AD在内的神经退行性疾病密切相关,在AD病理组织分析中常可发现MAO活性的异常升高。因此,抑制MAO的活性可以调节单胺类递质的代谢,维持其动态平衡,进而有利于增强AD患者的记忆功能。Lu等[28]将MAO抑制剂的活性片段引入到他克林分子中,合成得到一系列偶联物,其代表性化合物24对AChE、MAO-A和MAO-B的抑制活性均达到纳摩尔级,IC50分别为55.4、192.6和129.0nmol·L-1,是设计较为成功的多靶向抑制剂,很值得对其作进一步的构效关系研究。
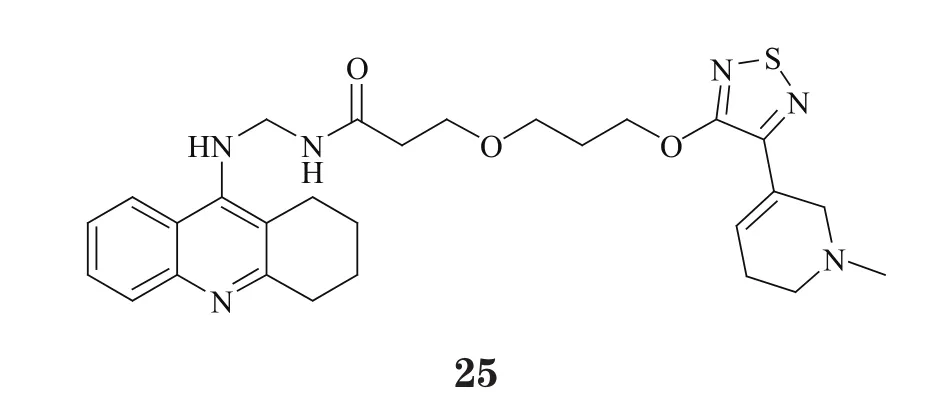
2.4.3 对毒蕈碱1型受体具有激动作用的多靶向他克林衍生物与MAO类似,毒蕈碱1型受体(M1受体)也是神经递质代谢及转运过程中的关键靶标之一,与AD密切相关。Fang等[29]将他克林与M1受体激动剂占诺美林(xanomeline)相结合,设计合成了一类全新的偶联物,其代表性化合物25在抑制AChE(IC50=7.9nmol·L-1)的同时,还能够显著激动M1受体(logKi=8.09)。
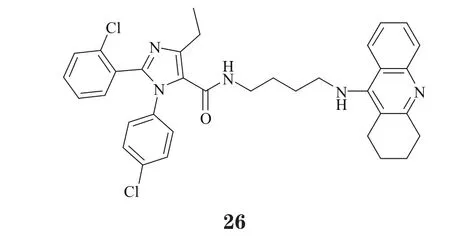
2.4.4 对大麻素受体具有抑制作用的多靶向他克林衍生物大麻素受体1(cannabinoidreceptor1,CB1)拮抗剂被认为在体内可提高ACh水平,从而发挥与AChE抑制剂的协同作用,其近年来已成为抗AD药物研发的热点。Lange等[30]将CB1活性片段与他克林相融合,设计出具有AChE和CB1双重抑制作用的化合物26,它对CB1具有高亲和力(Ki=48.0nmol·L-1),对AChE也保持了中等抑制活性(pIC50=6.5),且为进一步优化结构,设计更具活性的AChE和CB1双重抑制剂,提供了良好的先导化合物。
3 结语与展望
抗AD药物的研发历史已有数十年,由于AD的病理机制极为复杂,迄今未有能根治AD的药物问世,如何研发更具疗效的抗AD药物将是一个极具挑战性的课题。尽管如此,以作用于胆碱能系统的AChE抑制剂为代表的药物研究仍取得了长足进展,人们也在不断的实践中逐步认识到,只针对某一个靶点或环节的单靶向药物难以实现对整体疾病网络的持续性调节,而针对AD发病进程中的多重作用靶标,设计多靶向治疗药物,无疑更具优势和价值。
从治疗靶标的角度分析,AChE仍然是AD治疗的核心靶标之一,如何选择其他靶点加以合理组合,促使药物发挥“一药多靶”的协同作用,是抗AD药物分子设计成败的关键。在此前的抗AD药物研究中,科学家们选择更多的靶标是与Aβ生成和聚集、神经递质代谢等相关。然而,笔者认为,抗AD药物研发的思路和视野不应仅局限于“靶标”这些相对孤立的“点”上,而更应着眼于AD病理调控网络的“面”——神经微环境。
脑内神经元的生长及生物功能的正常发挥,有赖于其所处神经微环境的动态平衡。已有研究表明,早在神经元病变之前,AD病人脑部的正常神经微环境即已遭到破坏。一方面,这种被破坏的微环境将导致神经元功能受阻进而发生病变;另一方面,病变的神经元将进一步释放出大量的损伤性因子,加剧神经微环境的恶化,从而形成恶性循环。人类随着年龄的增长,其神经微环境中慢性炎症的发生明显增多,表现为,脑内氧化损伤增加,毒素清除能力减弱,免疫机能下降,最终形成微环境的不可逆损伤。目前已有许多研究认为,神经微环境的损伤很可能是AD病变早期阶段的首要诱因[31]。因此,如果能有效控制甚至逆转AD病人的神经微环境损伤,不仅能对AD的治疗起到决定性作用,更能在早期预防AD的发生,避免AD发病的低龄化趋势。就此角度而言,具有神经微环境调节作用的信号分子及通路,如保护性转录因子Nrf2、NF-κB、STAT3以及一些关键的表观调控机制,有可能成为新型抗AD药物研发的靶标方向。
从药物分子设计的角度分析,目前绝大多数的多靶向抗AD药物,是通过分子拼合策略,将数个针对不同靶标的活性片段整合而形成一个整体的分子骨架。由此获得的分子虽然具有较好的多靶向性,但其不可避免的存在如下弊端:1)分子结构较为复杂,不利于工业化制备和生产;2)类药性差,往往具有很大的相对分子质量和很高的脂溶性,极大地增加了后续开发的难度;3)活性片段间的连接链往往是一些较长的脂溶性侧链,而过高的脂溶性极易引起分子在蛋白表面的非特异性聚集,造成非特异性的靶标识别错误,从而导致脱靶效应;4)从本质上说,现有的多靶向抗AD药物的研发大多依赖于对已有药物结构的延伸,区别仅在于其组合方式的不同,不利于药物结构的创新。因此,笔者认为,应突破这种简单的拼合思路,立足于靶标的整体化学空间,选择对配体结合最为关键的区域进行药物靶向结构的研究与发现;同时,应以分子片段化的视角理解多靶向抗AD药物的结构,以获得结构多样化的全新活性片段,并以此为先导物,再采用分子融合策略对其加以整合和调配,化繁为简,从而克服现有多靶标药物分子的弊端。就此角度而言,现代药物设计手段,如基于碎片的药物设计、基于生物物理法的药物筛选及评价、基于信息学策略的药物发现等,将在多靶向抗AD药物的研发中,扮演越来越重要的角色。此外,许多天然产物如白藜芦醇、汉黄芩素、没食子酸等,经研究证实具有优良的神经微环境调节及保护作用,它们可通过还原脑部过度产生的超氧自由基、减缓氧化应激、增强还原性二项代谢酶的表达、调节蛋白的代谢平衡等机制而发挥良好的神经元保护作用。因此,若能对这些天然产物进行合理的利用和改造,提取有价值的结构片段融入多靶向分子的整体架构之中,也将为创新抗AD药物的研发提供更多的思路。
[1]Brookmeyer R, Johnson E, Ziegler-Graham K, et al. Forecasting the global burden of Alzheimer’s disease[J]. Alzheimers Dement, 2007, 3(3): 186-191.
[2]Rosini M, Simoni E,. Milelli A, et al. Oxidative stress in Alzheimer's disease: are we connecting the dots?[J]. J Med Chem, 2014, 57(7): 2821-2831.
[3]Pettersson M, Johnson D S, Subramanyam C, et al. Design, synthesis, and pharmacological evaluation of a novel series of pyridopyrazine-1,6-dione gamma-secretase modulators[J]. J Med Chem, 2014, 57(3): 1046-1062.
[4]Andersson C D, Forsgren N, Linusson A, et al. Divergent structure–activity relationships of structurally similar acetylcholinesterase inhibitors[J]. J Med Chem, 2013, 56(19): 7615-7624.
[5]León R, Garcia A G, Marco-Contelles J. Recent advances in the multitarget-directed ligands approach for the treatment of Alzheimer’s disease[J]. Med Res Rev, 2013, 33(1): 139-189.
[6]SchorNF.WhatthehaltedphaseIIIγ-secretaseinhibitortrialmay(ormay not) be telling us[J]. Ann Neurol, 2011, 69(2): 237-239.
[7]Torre J C. Phase 3 trials of solanezumab and bapineuzumab for Alzheimer's disease[J]. N Engl J Med, 2014, 370(15): 1459-1460.
[8]Fernandez-Bachiller M I, Perez C, Monjas L, et al. New tacrine-4-oxo-4H-chromene hybrids as multifunctional agents for the treatment of Alzheimer's disease, with cholinergic, antioxidant, and beta-amyloidreducing properties[J]. J Med Chem, 2012, 55(3): 1303-1317.
[9]Chen X, Zenger K, Decker M. Tacrine-silibinin codrug shows neuro- and hepatoprotective effects in vitro and pro-cognitive and hepatoprotective effects in vivo[J]. J Med Chem, 2012, 55(11): 5231-5242.
[10]Gemma S, Gabellieri E, Huleatt P, et al. Discovery of huperzine A-tacrine hybrids as potent inhibitors of human cholinesterases targeting their midgorge recognition sites[J]. J Med Chem, 2006, 49(11): 3421-3425.
[11]Rydberg E H, Brumshtein B, Greenblatt H M, et al. Complexes of alkylene-linked tacrine dimers with Torpedo californica acetylcholinesterase: Binding of Bis5-tacrine produces a dramatic rearrangement in the active-site gorge[J]. J Med Chem, 2006, 49(18): 5491-5500.
[12]Alonso D, Dorronsoro I, Rubio L, et al. Donepezil-tacrine hybrid related derivatives as new dual binding site inhibitors of AChE[J]. Bioorg Med Chem, 2005, 13(24): 6588-6597.
[13]Shao D, Zou C, Luo C, et al. Synthesis and evaluation of tacrine-E2020 hybrids as acetylcholinesterase inhibitors for the treatment of Alzheimer’s disease[J]. Bioorg Med Chem Lett, 2004, 14(18): 4639-4642.
[14]Munoz-Ruiz P, Rubio L, Garcia-Palomero E, et al. Design, synthesis,and biological evaluation of dual binding site acetylcholinesterase inhibitors: new disease-modifying agents for Alzheimer’s disease[J]. J Med Chem, 2005, 48(23): 7223-7233.
[15]Krasinski A, Radic Z, Manetsch R, et al. In situ selection of lead compounds by click chemistry: target-guided optimization of acetylcholinesterase inhibitors[J]. J Am Chem Soc, 2005, 127(18): 6686-6692.
[16]Fernández-Bachiller M I, Pérez C, Monjas L, et al. New tacrine-4-oxo-4H-chromene hybrids as multifunctional agents for the treatment of Alzheimer's disease, with cholinergic, antioxidant, and beta-amyloidreducing properties[J]. J Med Chem, 2012, 55(3): 1303-1317.
[17]Rizzo S, Bisi A, Bartolini M, et al. Multi-target strategy to address Alzheimer's disease: design, synthesis and biological evaluation of new tacrine-based dimers[J]. Eur J Med Chem, 2011, 46(9): 4336-4343.
[18]Sun Q, Peng D Y, Yang S G, et al. Syntheses of coumarin-tacrine hybrids as dual-site acetylcholinesterase inhibitors and their activity againstbutylcholinesterase,Aβaggregation,andβ-secretase[J].Bioorg Med Chem, 2014, 22(17): 4784-4791.
[19]Chen Y, Lehmann L, Zhang Y H, et al. Design, synthesis and evaluation of tacrine-flurbiprofen-nitrate trihybrids as novel anti-Alzheimer's disease agents[J]. Bioorg Med Chem, 2013, 21(9): 2462-2470.
[20]Fernández-Bachiller M I, Pérez C, González-Muñoz G C, et al. Novel tacrine-8-hydroxyquinoline hybrids as multifunctional agents for the treatment of Alzheimer's disease, with neuroprotective, cholinergic, antioxidant, and copper-complexing properties[J]. J Med Chem, 2010, 53(13): 4927-4937.
[21]Fang L, Kraus B, Lehmann J, et al. Design and synthesis of tacrineferulic acid hybrids as multi-potent anti-Alzheimer drug candidates[J]. Bioorg Med Chem Lett, 2008, 18(9): 2905-2909.
[22]Rosini M, Andrisano V, Mellor I R, et al. Inhibition of acetylcholinesterase, beta-amyloid aggregation, and NMDA receptors in Alzheimer's disease: a promising direction for the multi-target-directed ligands gold rush[J]. J Med Chem, 2008, 51(15): 4381-4384.
[23]Rosini M, Simoni E, Milelli A, et al. Oxidative stress in Alzheimer's disease: are we connecting the dots?[J]. J Med Chem, 2014, 57(7): 2821-2831.
[24]Thiratmatrakul S, Yenjai C, Waiwut P, et al. Synthesis, biological evaluation and molecular modeling study of novel tacrine-carbazole hybrids as potential multifunctional agents for the treatment of Alzheimer's disease[J]. Eur J Med Chem, 2014, 75: 21-30.
[25]Chen Y, Sun J, Lehmann J, et al. Tacrine-ferulic acid-nitric oxide (NO) donor trihybrids as potent, multifunctional acetyl- and butyrylcholinesterase inhibitors[J]. J Med Chem, 2012, 55(9): 4309-4321.
[26]Fang L, Peng S, Zhang Y, et al. Synthesis and biological evaluation of NO-donor-tacrine hybrids as hepatoprotective anti-Alzheimer drug candidates[J]. J Med Chem, 2008, 51(4): 713-716.
[27]Di Pietro O, Pérez-Areales F J, Juárez-Jiménez J, et al. Tetrahydrobenzo[h] [1,6]naphthyridine-6-chlorotacrine hybrids as a new family of anti-Alzheimer agents targeting beta-amyloid, tau, and cholinesterase pathologies[J]. Eur J Med Chem, 2014, 84: 107-117.
[28]Lu C, Zhou Q, Yan J, et al. A novel series of tacrine-selegiline hybrids with cholinesterase and monoamine oxidase inhibition activities for the treatment of Alzheimer's disease[J]. Eur J Med Chem, 2013, 62: 745-753.
[29]Fang L, Jumpertz S, Zhang Y, et al. Hybrid molecules from xanomeline and tacrine: enhanced tacrine actions on cholinesterases and muscarinic M1 receptors[J]. J Med Chem, 2010, 53(5): 2094-2103.
[30]Lange J H, Coolen H K, Kruse C G, et al. Design, synthesis, biological properties, and molecular modeling investigations of novel tacrine derivatives with a combination of acetylcholinesterase inhibition and cannabinoid CB1 receptor antagonism[J]. J Med Chem, 2010, 53(3): 1338-1346.
[31]Nunomura A, Castellani R J, Zhu X, et al. Involvement of oxidative stress in Alzheimer disease[J]. J Neuropathol Exp Neurol, 2006, 65(7): 631-641.
Progress in Novel Multi-target-directed Tacrine Derivatives
CHEN Yao1, SUN Haopeng2, LI Wei1
( 1. School of Pharmacy, Nanjing University of Traditional Chinese Medicine, Nanjing 210023, China; 2. School of Pharmacy, China Pharmaceutical University, Nanjing 210009, China)
Alzheimer’s disease (AD), one of the most common neurodegenerative diseases, not only threatens the health of elderly people, but alsoburdensthewholesocialmedicalandhealthsystem.Asaresult,thedesignanddevelopmentofeffcaciousandsafeanti-ADagentshasbecomeahotspotinthefeldofpharmaceuticalresearch.Tillnow,thecomplicatedpathogenesisofADhasnotbeenelucidated,indicatingthatthetreatmentonlyforasingletargetisdiffculttofullyachievetherapeuticeffects,andtherefore,themulti-target-directedagentsareconsideredtobethenewtrendfor the development of anti-AD agents. Acetylcholinesterase (AChE) inhibitors are the most successful compounds in the clinical treatment of AD at presenttime.Althoughtacrine,thefrstgenerationofAChEinhibitors,hasbeenwithdrawnduetoseveretoxicity,itsstructureissimple,itsactivityisclearanditshowsitssuperiorityinthestructuralmodifcationforthedesignofcomplicatedmulti-target-directedmolecules.Herefrom,tacrineastheideal active fragment could be introduced into the overall molecular skeletons of the multi-target-directed anti-AD agents. With some typical cases, the advances in the design of multi-target-directed anti-AD agents based on tacrine structure were reviewed and the design ideas were discussed so as to provide the reference for the further development of novel multi-target-directed tacrine derivatives.
Alzheimer’s disease; acetylcholinesterase inhibitor; tacrine; multi-target-directed derivative; molecular design
R971; R914.2
A
1001-5094(2014)09-0656-09
接受日期:2014-08-27
项目资助:南京中医药大学青年自然科学基金(No.13XZR21)
*通讯作者:李伟,研究员;
研究方向:天然药物化学及药物化学;
Tel : 025-86798269;E-mail: liwaii@126.com
猜你喜欢
世界科学技术-中医药现代化(2022年9期)2023-01-17
高中数理化(2022年2期)2022-02-22
军民两用技术与产品(2021年10期)2021-03-16
世界农药(2019年3期)2019-09-10
世界农药(2019年3期)2019-09-10
儿童故事画报·发现号趣味百科(2019年5期)2019-07-14
小天使·五年级语数英综合(2017年8期)2017-08-09
中学生数理化·高二版(2016年3期)2016-12-26
天然产物研究与开发(2016年11期)2016-06-15
肿瘤影像学(2015年3期)2015-12-09
