
海水中铁载体的固相萃取预处理和高效液相色谱
2015-09-11 07:09章蕾袁东星方锴刘宝敏
分析化学 2015年9期
章蕾+袁东星+方锴+刘宝敏

摘 要 铁载体是一种由海洋菌类合成并分泌、能特异性络合海水中铁的有机配体。本研究建立了固相萃取预处理、高效液相色谱-串联质谱测定海水中铁载体化合物的分析方法。海水样品经0.22 μm滤膜过滤,其中铁载体由ENVI-18萃取柱萃取、甲醇洗脱后得到富集净化后的试样; 采用SB-C18反向色谱柱以0.1%(V/V)甲酸溶液和甲醇溶液梯度洗脱分离,在质谱的多反应监测正离子模式下检测。3种铁载体化合物标准物质Pyoverdines-Fe, Ferrichrome, Ferrioxamine E的检测线性范围分别为0.001~3.0 μg/mL, 0.005~15.0 μg/mL, 0.001~3.0 μg/mL,相关系数R2>0.99; 仪器检出限分别为0.08, 1.76 和1.36 ng/mL; 定量限分别0.27, 5.87和4.53 ng/mL。在海水样品中添加铁载体化合物混合标准溶液,3种目标物测定值的相对标准偏差<12%,方法回收率分别为Pyoverdines-Fe 12.1%~18.6%,Ferrichrome 82.0%~97.7%,Ferrioxamine E 70.0%~98.3%。
关键词 铁载体; 固相萃取; 高效液相色谱-串联质谱; 海水
1 引 言
不同于地表水,海洋表层的生物可利用态铁(Fe)浓度极低,仅为nmol/L。铁载体是一种低分子量(0.5~1.5 kDa)的有机配体[1],对铁有极强的螯合能力。在铁限制条件下,铁载体由海洋中的一些细菌和真菌合成并分泌到细胞外,与海水中的铁特异性结合,通过细胞膜上特定铁载体受体的识别和转运,有效地将铁运输到微生物细胞内,满足生物体对铁的需求[1~3]。铁载体种类繁多,根据其官能团的不同,可分为氧肟酸型、酚-儿茶酚型和少量柠檬酸型。铁载体是海洋生物量的重要影响因子,其定性定量分析数据的获取是相关研究的关键。
由于铁载体结构复杂多样,在海洋环境中浓度低[4~6],分析相对困难,一般需要采集大量水样进行萃取和富集。传统的前处理方法,如液-液萃取法、柱层析法等[6~8],存在着操作繁琐、重现性较差等缺点; 而固相萃取法用于铁载体萃取[9,10]具有重现性好、溶剂用量小、操作高效快速等优点。测定铁载体常用分光光度法,如CAS(Chrome azurol S test)法[11]用于大多数细菌和真菌分泌的铁载体的检测。Csky等[12]特异性检测了氧肟酸型铁载体; Arnow建立了测定酚-儿茶酚型铁载体的方法[13]。上述方法易受基底干扰[1],回收率差,灵敏度低。Kilz等[14]尝试使用高效液相色谱-电喷雾离子源-质谱法(HPLC-ESI-MS)筛选荧光素类铁载体,该方法分析速度快,但仅用于定性分析。
已有的分析方法未能涵盖种类繁多的铁载体,且回收率均不佳。据文献[9,15]报道,天然海水中能被检测到的铁载体均为氧肟酸型铁载体,而含儿茶酚官能团的铁载体可能因亲脂性相对较高,未能在水相中检测得到。本研究旨在建立天然海水中铁载体的分析方法,故选择氧肟酸型铁载体为研究对象,以固相萃取法对海水样品进行预处理,以高效液相色谱-串联质谱(LC-MS/MS)测定。本方法操作简便、基底干扰小,回收率优于已报道的,并能提供目标物的结构信息,适于海水基底中铁载体的分析。
2 实验部分
2.1 仪器与试剂
Agilent6410 Triple Quad液相色谱-串联质谱联用仪(LC-MS/MS,美国Agilent公司),配Agilent 1290液相色谱系统、电喷雾离子源(ESI); Milli-Q超纯水机(美国Millipore公司); 0.22 μm聚醚砜针式过滤器、0.22 μm聚醚砜滤膜(美国Pall公司)。
甲醇(色谱纯,美国Tedia公司); 甲酸(96%,ACS纯,美国Sigma公司); 乙腈(色谱纯, 美国Tedia公司); NaCl(优级纯,国药集团化学试剂有限公司); HCl(工艺超纯,昆山金城试剂有限公司); NaOH(ACS试剂,美国Sigma公司); 固相萃取柱:500 mg/6 mL Supelclean LC-18(美国Supelco公司),200 mg/6 mL Waters OASIS HLB(美国Waters公司),100 mg/1 mL Isolute ENV+(瑞典Biotage公司),500 mg/6 mL Supelclean ENVI-18、1 g/6 mL Supelclean ENVI-Carb(美国Supelco公司)。
本研究选择的3种氧肟酸型铁载体均购自美国Sigma公司:Ferrichrome(FC,无铁络合基,货号F8014)、Ferrioxamine E(FO E,含铁络合物,货号38266)、Pyoverdines-Fe(PVDs-Fe,含铁络合物,货号P8374),其对应的结构式示于Sigma公司的产品说明书。FC和FO E呈环状,是典型且常见的氧肟酸型铁载体化合物; PVDs-Fe呈链状,因其官能团多样,为混合型铁载体化合物,既含氧肟酸官能团,又有儿茶酚官能团。3种铁载体均以1∶1比例与Fe络合。
2.2 溶液配制
铁载体化合物标准储备溶液: 超纯水配制,浓度分别为PVDs-Fe,200 μg/mL; FC,500 μg/mL; FO E,200 μg/mL; 于
20℃下保存。使用时用超纯水配制成所需浓度。低浓度: PVDs-Fe 0.01 μg/mL、FC 0.05 μg/mL、FO E 0.01 μg/mL; 中浓度: PVDs-Fe 0.10 μg/mL、FC 0.50 μg/mL、FO E 0.10 μg/mL; 高浓度: PVDs-Fe 1.00 μg/mL、FC 5.00 μg/mL、FO E 1.00 μg/mL。
含铁载体化合物的人工海水: 取1~10 L超纯水,用NaCl调节至所需盐度,用0.01 mol/L HCl和0.01 mol/L NaOH调节至所需pH值,向水样中加入1 mL铁载体混合标准溶液,其中各目标物含量分别为 PVDs-Fe 3.00 μg/mL、FC 15.00 μg/mL、FO E 3.00 μg/mL。
2.3 样品预处理
天然海水样品经0.22 μm滤膜过滤后,
20℃保存,使用前解冻,人工海水样品可不过滤。分别用5 mL甲醇、5 mL超纯水活化ENVI-18固相萃取柱,以5 mL/min的流速使水样通过萃取柱。用5 mL超纯水预淋洗萃取柱,以4 mL甲醇洗脱目标物,将收集的洗脱液氮吹至近干(小于100 μL),用含0.1%(V/V)甲酸定容至1.00 mL,涡旋混匀后经0.22 μm针式过滤器过滤,待测。
2.4 色谱和质谱分析液相色谱参数: ZORBAX SB-C18色谱柱(50 mm×2.1 mm,1.8 μm,美国Agilent公司); 进样量: 10 μL; 流动相: A为0.1%(V/V)甲酸,B为甲醇; 流动相流速: 0.25 mL/min; 梯度程序: 0~3 min,5% B; 3~18 min,5%~100% B; 18~20 min,100% B; 20~20.01 min,100%~5% B; 20.01~23 min,5% B。
质谱参数: 电喷雾离子源(ESI); 多反应监测(MRM)模式; 毛细管电压: 2886 V; 鞘气温度: 200℃; 鞘气流速: 6 L/min; 干燥气温度: 300℃; 干燥气流速: 10 L/min; 雾化气压力: 240.28 kPa; 喷嘴电压: 1500 V; 碰撞气为高纯氮气(纯度>99.999%)。以ESI+模式,在m/z 100~1400范围内选择合适的区间进行全扫描,选择准分子离子峰; 再以准分子离子峰作为母离子进行二级质谱扫描,选择准分子离子和两个信号强度适宜的子离子作为监测反应的离子对。优化后目标物的质谱分析参数列于表1。
3 结果与讨论
3.1 色谱条件的优化
文献[9]表明,经典的C18反向色谱法分离铁络合物时易形成拖尾峰,目标物的出峰时间与溶剂峰接近或重合。选择合适的流动相可抑制拖尾。实验以3种铁载体化合物的分离效果和信号强度为指标,评估了4种流动相: (1)甲醇-超纯水、(2)甲醇-0.1%(V/V)甲酸、(3)乙腈-超纯水、(4)乙腈-0.1%(V/V)甲酸。
结果表明,采用4种流动相均可实现对目标物的分离。在流动相中加入甲酸,铁载体化合物的峰形及信号响应得到明显改善,而流动相(2)的分离结果最优。本实验选择流动相(2),采用梯度洗脱模式。LC-MS/MS分析中常用的甲酸含量为0.1%(V/V),本实验未对流动相中甲酸含量做进一步研究。优化后, PVDs-Fe,FC和FO E的保留时间分别为5.46,7.70和8.43 min。
3.2 固相萃取条件的优化
天然海水样品中铁载体浓度低,需富集。近年来,固相萃取技术在样品预处理领域运用广泛,McCormack等[9]用Isolute ENV+固相萃取柱,对人工及实际海水中铁载体进行萃取; 而后,该方法被沿用于海水中铁载体的萃取[10,15~17]。但该方法的回收率不高,人工海水中的铁载体化合物回收率为Fe-rhodotoluate,7.9%; Ferrioxamine,46%; FC,48%; 实际海水样品中目标物的回收率更低,Fe-rhodotoluate,4.0%; Ferrioxamine,21%; FC,37%。
本研究系统考察了固相萃取柱(见2.1节)、洗脱溶剂中甲醇的比例(甲醇-超纯水(V/V): 0%, 30%, 50%, 80%和100%)、洗脱溶剂用量(1.00, 2.00, 3.00, 4.00和5.00 mL)、上样体积(1.0, 3.0, 5.0, 8.0和10.0 mL)、盐度(0‰, 15‰, 25‰, 35‰和40‰)、pH值(6.50, 7.00, 7.50, 8.00, 8.50)等参数对萃取结果的影响,通过正交实验优选参数。实验采用六因素五水平25次实验,记为L25(56),以各目标物的回收率为评估依据,选出最优固相萃取条件。
根据极差分析,6种因素中,对铁载体化合物萃取回收率影响最大的是洗脱溶剂的比例。在正交实验范围内,综合3种目标物回收率情况,选择较好的萃取条件为: Supelclean ENVI-18柱萃取,4.00 mL 100%甲醇洗脱,上样体积1.0 L,盐度35‰,pH 7.00。在此条件下,回收率为PVDs-Fe 34.46%, FC 96.29%, FO E 86.11%。上样体积对PVDs-Fe回收率的影响较大,而FC和FO E的回收率在水样少于10 L时基本不变。本研究又对pH值进行单因素考察,结果表明,在pH 7.02~8.52范围内,目标物回收率基本持平。全球海洋表层海水盐度大多介于33‰~37‰之间,表层水的pH值介于7.9~8.4之间[18],故本方法可用于海水中铁载体化合物的直接萃取,无需调节海水样品的盐度及pH值。
3.3 基质效应的影响
实验考察了LC-MS/MS分析的基质效应的影响。以厦门近岸海水为基底,按2.3节方法进行过滤和预处理,氮吹浓缩至干,分别用高、低2个浓度水平的铁载体化合物混合标准溶液溶解并定容。分析此溶液和用2.2节方法配制的相同浓度的混合标准纯水溶液,比较其响应,基质效应=(海水样品的信号响应/纯水样品的信号响应)×100%[19]。基质效应在80%~120%之间表示效应不明显,高于120%为正效应,反之为基质抑制效应。结果表明,本方法的基质效应在97.5%~124.4%范围内,仅低浓度FC的超出120%,表现出轻微正效应。后续实验采用非基质匹配的标准工作曲线进行定量分析。
3.4 质谱分析
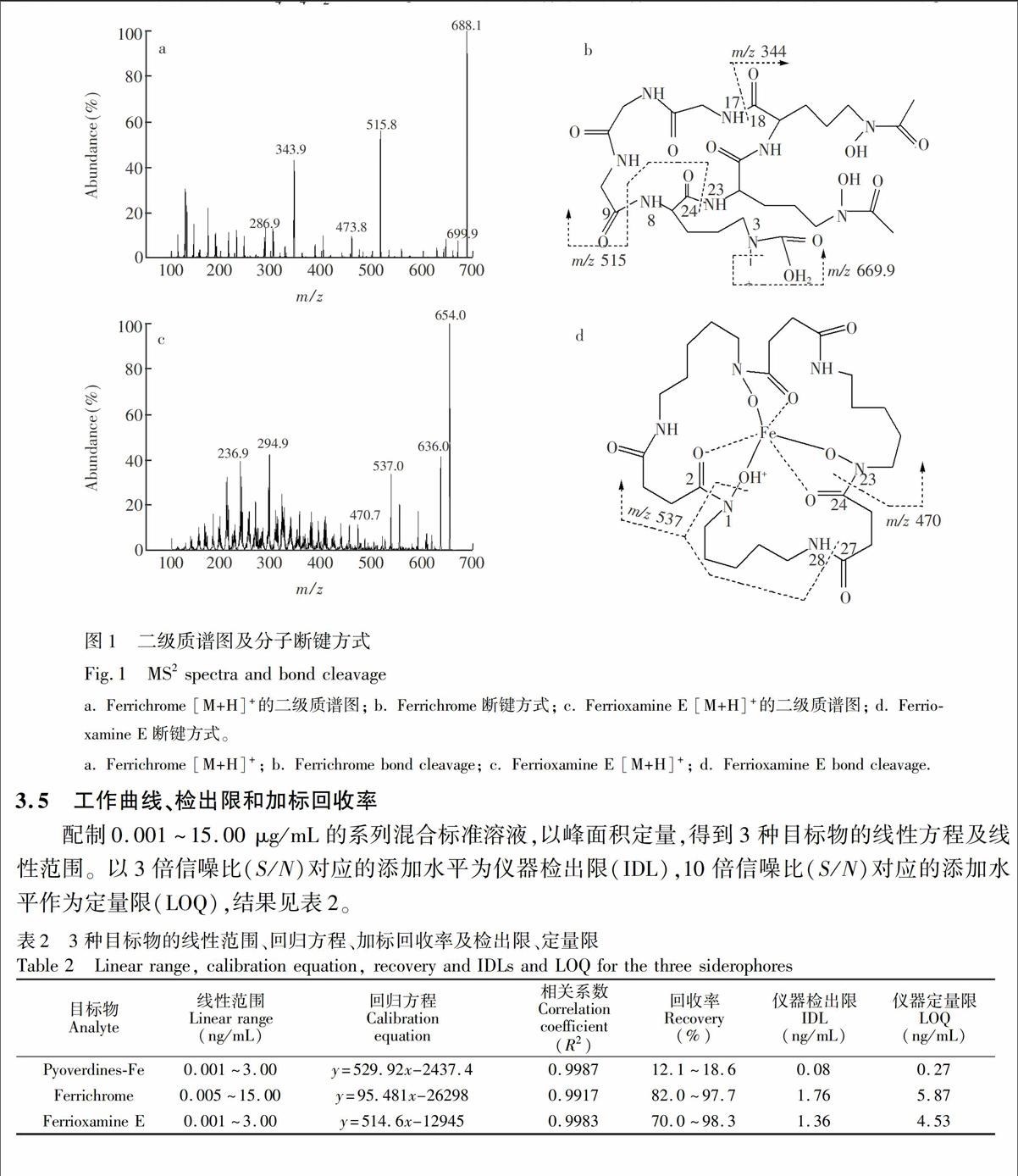
实验以甲醇-水(50∶50,V/V)为载流,流速0.25 mL/min,不接色谱柱,单标进样10 μL。在电喷雾电离过程中,3种目标物所形成的主导离子均为质子化的母体,且在ESI+模式下有较强的信号响应,其中FC和FO E有[M+H]+和[M+Na]+准分子离子峰。PVDs-Fe为3种Pyoverdine的混合物,结合其分子量信息及质谱图可获PVDs-Fe 的[M+2H]2+及[M+H]+准分子离子峰; 混合物中以含琥珀酸和琥珀酰胺的Pyoverdines为主要成分,含2-羟基戊二酰亚胺结构的Pyoverdine为次要成分。将获得的母离子进行二级质谱扫描,可分别获得其碎片离子信息(图1),用于铁载体化合物谱图的初步解析。本研究仅对FC和FO E进行MS2分析。
如图1b所示, 后易脱H2O生成m/z 669.9的产物离子,丰度较大的碎片离子m/z 516可通过8-9和23-24两处酰胺键的断裂形成,经过进一步的断裂(17-18位酰胺键)可得到m/z 344的碎片离子。FO E是Ferrioxamine族中仅有的两个具有环形结构(Ferrioxamine E和D2)的成员之一,其它Ferrioxamine均为链状结构[1]。链状结构的Ferrioxamines MS2谱中可观察到因链末端脱-NH2或-NH3而产生的碎片离子峰[20,21],而环状结构的Ferrioxamines MS2谱中可观察到因脱H2O而产生的碎片离子,如FO E 产生的m/z 636。质子化的FO E的质谱图文献[21]报道过,本实验结果与之相符,但由于所用的检测器不同,所得FO E的各碎片离子峰丰度略有差异。本实验对丰度较大的碎片离子进行谱图解析,见图1d。 FO E的分子离子峰为m/z 654。 m/z 554来源于1位NO键的断裂,1-2位NC键的断裂及27-28位酰胺键的断裂。m/z 554经进一步碎裂,丢失OH基团后可得碎片离子m/z 537; m/z 470.7的产生则源于m/z 554碎片离子中C4H4O2的丢失。建立铁载体谱库是质谱技术用于定性分析的重要条件。
3.5 工作曲线、检出限和加标回收率
配制0.001~15.00 μg/mL的系列混合标准溶液,以峰面积定量,得到3种目标物的线性方程及线性范围。以3倍信噪比(S/N)对应的添加水平为仪器检出限(IDL),10倍信噪比(S/N)对应的添加水平作为定量限(LOQ),结果见表2。
以厦门近岸海水为基底,按2.3节方法过滤后,分别向500 mL海水中加入低、中、高3个浓度水平的铁载体化合物混合标准溶液各1 mL。每个浓度做4份平行,以标准工作曲线法进行定量分析,考察方法的回收率及其相对标准偏差(RSD)。3种目标物的加标回收率为PVDs-Fe 12.1%~18.6%,FC 82.0%~97.7%,FO E 70.0%~98.3%。PVDs-Fe的回收率较低,但该化合物的回收率为首次报道,与同类研究的其它铁载体回收率[9]相比属于常规水平,而FC和FO E的回收率均高于报道值[9,10]。3种目标物回收率的RSD分别为PVDs-Fe 12.5%,FC 3.9%,FO E 8.9%。本方法对络合态铁载体化合物PVDs-Fe、FO E和非络合铁载体FC均具有良好的定性与定量分析能力。
3.6 实际海水样品分析
运用本方法测定厦门鼓浪屿附近海水中铁载体化合物含量。取样量分别为1, 5和10 L,均未检出铁载体或类铁载体化合物,这与近岸海水中铁的浓度较高有关。本课题组对邻近鼓浪屿的九龙江河口海域的监测结果[22]表明,该海域溶解态铁含量为15.7~48.8 μg/L,此量级与可检出铁载体海域如新西兰东南部沿海地区的溶解态铁含量0.22~0.45 nmol/L [6]相比,属较高水平,不足以构成铁胁迫,无法刺激生物分泌铁载体。
4 结 论
建立了适用于海水基底中铁载体及其铁络合物分离鉴定的固相萃取预处理、LC-MS/MS测定法。方法的基底分离效果好、富集倍数高,回收率良好,定量分析准确; 利用质谱技术还能对目标物的结构进行定性分析。本方法适合于高营养盐低叶绿素等铁限制条件的海区中海水样品的测定。
References
1 Winkelmann G. CRC Handbook of Microbial Iron Chelates. CRC: Florida, 1991: 15-64
2 Perry R D,Bobrov A G, Fetherston J D. Metallomics, 2015 (doi:10.1039/C4MT00332B)
3 Chu B C, Garcia-Herrero A, Johanson T H, Krewulak K D, Lau C K, Peacock R S, Slavinskaya Z, Vogel H J. Biometals, 2010, 23(4): 601-611
4 Rue E L, Bruland K W. Mar. Chem., 1995, 50(1): 117-138
5 Gledhill M, Buck K N. Front. Microbiol., 2012, 3: 1-17
6 Velasquez I, Nunn B L, Ibisanmi E, Goodlett D R, Hunter K A, Sander S G. Mar. Chem., 2011, 127(1): 97-107
7 Armstrong J E, Van Baalen C. J. Gen. Microbiol., 1979, 111(2): 253-262
8 Jalal M A, Mocharla R, Barnes C L, Hossain M B, Powell D R, Eng-Wilmot D L, Grayson S L, Benson B A, van der Helm D. J. Bacteriol., 1984, 158(2): 683-694
9 McCormack P, Worsfold P J, Gledhill M. Anal. Chem., 2003, 75(11): 2647-2652
10 Mawji E, Gledhill M, Milton J A, Tarran G A. Environ. Sci. Technol., 2008, 42(23): 8675-8680
11 Schwyn B, Neilands J B. Anal. Biochem., 1987, 160(1): 47-56
12 Csáky T Z, Hassel O, Rosenberg T, Turunen E, Tuhkanen A. Acta Chem. Scand., 1948, 2: 450-454
13 Arnow L E. J. Biochem., 1937, 118(2): 531-537
14 Kilz S, Lenz C, Fuchs R, Budzikiewicz H. J. Mass Spectrom., 1999, 34(4): 281-290
15 Mawji E, Gledhill M, Milton J A, Zubkov M V, Thompson A, Wolff G A, Achterberg E P. Mar. Chem., 2011, 124(1): 90-99
16 Gledhill M, McCormack P, Ussher S, Achterberg E P, Mantoura R F C, Worsfold P J. Mar. Chem., 2004, 88(1): 75-83
17 Moberg M, Nilsson E M, Holmstrm S J, Lundstrm U S, Pettersson J, Markides K E. Anal. Chem., 2004, 76(9): 2618-2622
18 CHEN Min. Chemical Oceanography. Beijing: Ocean Press, 2009: 22-73
陈 敏. 化学海洋学, 北京: 海洋出版社, 2009: 22-73
19 Matuszewski B K, Constanzer M L, Chavez-Eng C M. Anal. Chem., 2003, 75(13): 3019-3030
20 Groenewold G S, Van Stipdonk M J, Gresham G L, Chien W, Bulleigh K, Howard A. J. Mass Spectrom., 2004, 39: 752-761
21 Mawji E, Gledhill M, Worsfold P J, Achterberg E P. Rapid Commun. Mass Spectrom., 2008, 22(14): 2195-2202
22 PENG Yuan-Zhen, HUANG Yong-Ming, YUAN Dong-Xing, LI Yan, GONG Zhen-Bin. Chinese J. Anal. Chem., 2012, 40(16): 877-882
猜你喜欢
中国中药杂志(2016年21期)2017-02-16
中国中药杂志(2016年21期)2017-02-16
分析化学(2017年1期)2017-02-06
热带农业科学(2016年10期)2016-12-12
分析化学(2016年7期)2016-12-08
分析化学(2016年7期)2016-12-08
科学与财富(2016年28期)2016-10-14
肉类研究(2015年1期)2015-04-08
