
4-磺酰胺-L-脯氨酸衍生物的合成及其水相中不对称催化Aldol反应的性能
2016-05-12 00:55刘玉霞吕名秀赵朋飞
化学研究 2016年2期
刘玉霞,吕名秀,卢 奎,赵朋飞
(河南工程学院 材料与化学工程学院, 河南 郑州 450007)
4-磺酰胺-L-脯氨酸衍生物的合成及其水相中不对称催化Aldol反应的性能
刘玉霞,吕名秀,卢奎,赵朋飞
(河南工程学院 材料与化学工程学院, 河南 郑州 450007)
摘要:以4-羟基-L-脯氨酸为原料合成了4-对甲基苯磺酰胺-L-脯氨酸催化剂1a-b,通过IR,1H NMR和HR MS对其结构进行了表征,并考察了其水相中对不对称Aldol反应的催化性能. 结果表明两种催化剂在水相中均能很好的催化Aldol反应,仅用5 mol%的催化剂即可得到很好的催化效果,产率高达93%,非对映选择性高达94∶6,而对映选择性高达99%.
关键词:脯氨酸衍生物;不对称催化;Aldol反应;水相催化
Received date: 2015-11-18.
Foundation item: The Natural Science Foundation of Henan province (grants 2011B150005) and The Doctoral Foundation of Henan Institute of Engineering (D09002).
Biography: LIU Yuxia (1979-), female, associate professor,research interest in organic synthesis.*Corresponding author, E-mail:liuyuxia_41@126.com
The asymmetric Aldol reaction catalized by organocatalysts is one of the most powerful methods for the construction of complex chiral polyol architectures. Since the discovery ofL-proline catalyzed intermolecular Aldol reactions, asymmetric organocatalysis has received growing attention[1]. A lot of organocatalysts have been synthesized and applied for highly enantioselective direct Aldol and othter reactions in the past few years[2-11]. Much effort has been devoted to developing 4-substituted-L-proline organocatalysts[12-13]. Stereoselective reaction in water/aqueous media is another important research area because water is an environmentally safe media, which avoids the problems of pollution that are inherent with organic solvents[14-15]. Herein, we reported two diastereomers 4-sulfonamido-L-proline 1a-b catalyzed the asymmetric Aldol reaction between aromatic aldehydes and cyclohexanone in water. These catalysts are capable of catalyzing the direct Aldol reaction in water at high enantiostereoselectivity (up to 93% yield and 99%ee).
1Experimental section
1.1General
All chemicals were used as received unless otherwise noted. Reagent grade solvents were redistilled prior to use. All1H NMR spectra were collected on a Bruker DPX 400 NMR spectrometer with TMS as an internal reference. FT-IR spectra were determined on a Thermo Nicolet IR200 unit. High resolution mass spectra (HR-MS) were obtained on a Waters Micromass Q-Tof MicroTMinstrument using the ESI technique. Chromatography was performed on silica gel (200-300 mesh). Melting points were determined by a XT5A apparatus and uncorrected. Optical rotations were determined on a Perkin Elmer341 polarimeter. Enantiomer excess was measured by chiral HPLC at room temperature using JASCO PU-1580 pump equipped with JASCO UV-1575 ultra detector (or Syltech 500 pump equipped with a UV 500 version 4.1 ultra-violet detector).
1.2Preparation of the catalysts 1a-b
1.2.1(2S, 4S)-1-benzyloxycarbonyl-2-methoxycarbonyl-4-chloro-pyrrolidine
To a stirred solution of N-Cbz-4-hydroxy -L-proline methyl ester 2 (3.4 g, 12.2 mmol) in pyridine (20 mL) was addedp-toluenesulfonyl chloride (2.78 g, 14.6 mmol) at room temperature. After stirring for about 12 h, the mixture was heated for 10 h at 60 ℃. The reaction mixture was concentrated under reduced pressure. The resulted residue was neutralized with 1 mol/L HCl and extracted with ethyl acetate. The organic layer was dried (Na2SO4), filtered and concentrated followed by column chromatography on silica gel (ethyl acetate /Petroleum ether = 1∶3,V/V) to give semi-solid product.
Yield 78%, IR (KBr, cm-1): 2 952, 1 755, 1 709, 1 415, 1 352, 1 266, 1 206, 1 166, 1 114, 1 034, 766, 699, 610; [α]D20: -36.8°, (c1.27, EtOH);1H NMR (400 MHz, CDCl3)δ: 2.40-2.45 (m, 1H, H-3), 2.70-2.74 (m, 1H, H-3), 3.64-3.78 (s, 3H, OCH3), 3.71-3.73 (m, 1H, H-5), 3.99-4.04 (m, 1H, H-5), 4.38-4.41 (m, 1H, H-2), 4.46-4.53 (m, 1H, H-4), 5.06-5.22 (m, 2H, -OCH2Ph), 7.31-7.37 (m, 5H, ArH); HR-MS (ESI):m/zcacld. for C14H17NO4Cl (M+H)+298.084 6, found 298.083 8. 1.2.2(2S, 4R)-1-benzyloxycarbonyl-2-methoxycarbonyl-4-(4′-methyl-benzene sulfonyl oxy)-pyrrolidine
To a stirred solution of N-Cbz-4-hydroxy-L-proline methyl ester 2 (3.4 g, 12.2 mmol) in pyridine (20 mL) was addedp-toluenesulfonyl chloride (2.78 g, 14.6 mmol) at room temperature. After stirring for about 12 h, the reaction mixture was concentrated under reduced pressure. The resulted residue was neutralized with 1 mol/L HCl and extracted with ethyl acetate. The organic layer was dried (Na2SO4), filtered and concentrated followed by column chromatography on silica gel (ethyl acetate /Petroleum ether = 1∶2,V/V) to give semi-solid product, which was recrystallized from ethanol as a colorless solid.
Yield 88%, m.p. 67-68 ℃, IR (KBr, cm-1): 3 412, 3 036, 2 956, 1 759 1 713, 1 597, 1 450, 1 419, 1 364, 1 266, 1 199, 1 178, 1 117, 1 054, 952, 905, 890, 753, 740, 555;1H NMR (400 MHz, CDCl3)δ: 2.15-2.21 (m, 1H, H-3), 2.41-2.59 (m, 1H, H-3), 2.41 and 2.43 (s, 3H, -Ar′CH3), 3.50 and 3.71 (s, 3H, -OCH3), 3.60-3.75 (m, 2H, H-5), 4.44-4.47 (m, 1H, H-2), 4.96-5.19 (m, 3H, H-4 and -OCH2Ph), 7.33-7.38 (m, 5H, Ph, 2H, Ar′), 7.73-7.78 (m, 2H, Ar′).1.2.3General procedure for the preparation of 4a-b
A solution of compound 3 (5.0 mmol) and sodium azide (20.0 mmol) in DMF(25 mL) was stirred at 60-70 ℃ for 2 h. 25 mL of water was added to the reaction mixture. The resulted solution was extracted with ethyl acetate. The organic layer was dried (Na2SO4), filtered and concentrated. The residue would be used without any further purification, yield 95%.
(2S, 4R)-1-benzyloxycarbonyl-2-methoxycarbonyl-4-triazo-pyrrolidine (4a) IR (KBr, cm-1): 3 426, 2 956, 2 108, 1 754, 1 709, 1 576, 1 452, 1 391, 1 204, 1 191, 1 133, 1 051, 772, 694, 567;1H NMR (400 MHz, CDCl3)δ: 2.22-2.25 (m, 1H, H-3), 2.43-2.47 (m, 1H, H-3), 3.57-3.60 (m, 1H, H-5), 3.77-3.82 (m, 1H, H-5), 3.64 and 3.77 (s, 3H, -OCH3), 4.19 (m, 1H, H-4), 4.43-4.52 (m, 1H, H-2), 5.05-5.21 (m, 2H, -OCH2Ph), 7.26-7.37 (m, 5H, Ph); MS (ESI):m/zcacld. for C14H17N4O4(M+H)+305.1, found 304.6.
(2S, 4S)-1-benzyloxycarbonyl-2-methoxycarbonyl-4-triazo-pyrrolidine (4b)IR (KBr, cm-1): 3 426, 2 956, 2 108, 1 754, 1 709, 1 576, 1 452, 1 391, 1 204, 1 191, 1 133, 1 051, 772, 694, 567;1H NMR (400 MHz, CDCl3)δ: 2.18-2.44 (m, 2H, H-3), 3.56 and 3.78 (s, 3H, -OCH3), 3.58-4.10 (m, 2H, H-5), 4.21-4.24 (m, 1H, H-4), 4.38-4.51 (m, 1H, H-2), 5.01-5.22 (m, 2H, -OCH2Ph), 7.29-7.37 (m, 5H, -Ph); MS (ESI):m/zcacld. for C14H17N4O4(M+H)+305.1, found 304.6.1.2.4General procedure for the preparation of 5a-b
A solution of compound 4 (1.45 g, 5.0 mmol), triphenylphosphine (2.62 g, 10.0 mmol) and water (0.18 mL, 10 mmol) in THF(25 mL) was refluxed for 6 h. Then the reaction mixture was concentrated, added 0.1 mol/L HCl, then filtered. The filtrate was neutralized to pH>8 with Na2CO3. The resulted solution was extracted with ethyl acetate. The organic layer was dried (Na2SO4), filtered and concentrated, yield 86%.
(2S, 4R)-1-benzyloxycarbonyl-2-methoxycarbonyl-4-amino-pyrrolidine (5a) IR (KBr, cm-1): 3 374, 3 386, 3 033, 2 952, 2 886, 1 747, 1 704, 1 498, 1 418, 1 356, 1 204, 1 170, 1 112, 769, 751, 699; [α]D20:43.0 (c1.34,EtOH);1H NMR (400 MHz, CDCl3)δ: 1.99-2.19 (m, 2H, H-3), 3.21-3.22 (m, 1H, H-4), 3.56-3.76 (s, 3H, -OCH3), 3.69-3.81 (m, 2H, H-5), 4.44-4.51 (m, 1H, H-2), 5.01-5.21 (m, 2H, -OCH2Ph), 7.27-7.36 (m, 5H, Ph); HR-MSm/z: calcd. For C14H19N2O4(M+H)+279.134 5, found 279.133 6.
(2S, 4S)-1-benzyloxycarbonyl-2-methoxycarbonyl-4-amino-pyrrolidine (5b)IR (KBr, cm-1): 3 374, 3 386, 3 033, 2 952, 2 886, 1 747, 1 704, 1 498, 1 418, 1 356, 1 204, 1 170, 1 112, 769, 751, 699; [α]D20: -23.2° (c1.18, EtOH);1H NMR (CDCl3, ppm): δ = 1.83-1.89 (1H, m, H-3), 2.42-2.48 (1H, m, H-3), 3.31-3.37 (1H, m, H-5), 3.56-3.57 (1H, m, H-5), 3.70-3.79 (1H, m, H-4), 3.59 and 3.77 (3H, s, -OCH3), 4.33-4.41 (1H, m, H-2), 5.02-5.20 (2H, m, -OCH2Ph), 7.28-7.37 (5H, m, Ph); HR-MSm/z: calcd. For C14H18N2O4(M+H)+279.134 5, found 279.133 3.
1.2.5General procedure for the preparation of 6a-b
To a stirred solution of 5 (0.84 g,3.0 mmol) and triethylamine (2 mL) in dichloromethane (10 mL) was addedp-toluenesulfonyl chloride (0.86 g,4.5 mmol) at room temperature. After stirring for about 3 h, evaporating solvent, ethyl acetate and 1 mol/L HCl was added. The organic layer was washed to neutral, dried (Na2SO4), filtered and concentrated. The residue could be used without any further purification, yield 95%.
(2S, 4R)-1-benzyloxycarbonyl-2-methoxycarbonyl-4-(4′-methylphenylsulfonamido)-pyrrolidine (6a) IR (KBr, cm-1): 3 254, 3 033, 2 954, 1 747, 1 708, 1 420, 1 352, 1 209, 1 161, 1 091, 914, 766, 699, 667;1H NMR (400 MHz, CDCl3)δ: 2.09-2.24 (m, 2H, H-3), 2.41 and 2.43 (s, 3H, -ArCH3), 3.24-3.28 (m, 1H, H-5), 3.62-3.69 (m, 1H, H-5), 3.55 and 3.71 (s, 3H, -OCH3), 3.92-3.96 (m, 1H, H-4), 4.34-4.42 (m, 1H, H-2), 4.96-5.16 (m, 3H, -OCH2Ph and TsNH-), 7.25-7.37 (m, 7H, -Ph and Ar′), 7.72-7.743 (d, 2H,J=6.4, Ar′).
(2S, 4S)-1-benzyloxycarbonyl-2-methoxycarbonyl-4-(4′-methylphenylsulfonamido)-pyrrolidine (6b) IR (KBr, cm-1): 3 261, 3 033, 2 954, 1 748, 1 708, 1 419, 1 355, 1 209, 1 162, 1 091, 914, 768, 700, 666;1H NMR (400 MHz, CDCl3)δ: 1.82-1.97 (m, 1H, H-3), 2.27-2.38 (m, 1H, H-3), 2.40 and 2.43 (s, 3H, -ArCH3), 3.39-3.57 (m, 2H, H-5), 3.60 and 3.77 (s, 3H, -OCH3), 4.03 (m, 1H, H-4), 4.27-4.32 (m, 1H, H-2), 4.99-5.16 (m, 2H, -OCH2Ph), 5.69-5.85 (m, 1H, TsNH-), 7.29-7.36 (m, 7H, Ph and Ar′), 7.71-7.73 (d, 2H,J=8.0, Ar′).1.2.6General procedure for the preparation of 1a-b
Compound 4 was dissolved in 10 mL of THF, and 5 mL of 10% NaOH aqueous solution was then added. The reaction mixture was stirred at room temperature for 5 h and acidified to pH <3 with 1 mol/L HCl. The resulted solution was extracted with ethyl acetate. The organic layer was dried (Na2SO4), filtered and concentrated. The residue was dissolved in 150 mL ethanol and 20% Pd/C (0.1 g) was added. After catalytic hydrogenation for 4 h at 3 atm, the mixture was concentrated to give semi-solid product, which was recrystallized from ethanol to furnish 1 as a colorless solid.
(2S, 4R)-4-(4′-methylphenylsulfonamido)-L-proline (1a)m.p. 244-246 ℃; [α]D20: -28.7°, (c0.52, EtOH); IR (KBr, cm-1): 3 416, 3 147, 2 922, 1 625, 1 448, 1 389, 1 331, 1 159, 1 091, 816, 713, 670;1H NMR (400 MHz, CDCl3)δ: 1.98 (m, 1H, H-3), 2.35 (m, 1H, H-3), 2.40 (s, 3H, -ArCH3), 2.71 (m, 1H, H-5), 3.18 (m, 2H, H-5), 3.61 (m, 1H, H-4), 3.74 (m, 1H, H-2); HR-MS (ESI):m/zcacld. for C12H17N2O4S (M+H)+307.072 3, found 307.072 5.
(2S, 4S)-4-(4′-methylphenylsulfonamido)-L-proline (1b)m.p. 261-262 ℃; [α]D20: +8.4°, (c0.52, EtOH); IR (KBr, cm-1): 3 416, 3 147, 2 922, 1 625, 1 448, 1 389, 1 331, 1 159, 1 091, 816, 713, 670;1H NMR (400 MHz, CDCl3)δ: 1.90 (m, 2H, H-3), 2.39 (s, 3H, -CH3), 2.83 (m, 1H, H-5), 3.20 (m, 1H, H-5), 3.52 (m, 1H, H-4), 3.70 (m, 1H, H-2); HR-MS (ESI):m/zcacld. for C12H17N2O4S (M+H)+307.072 3, found 307.071 6.
1.3General procedure for the Aldol condensations
0.33 mmol of aldehyde was added to a mixture of 0.3 mL of ketone, 0.7 mL of water and 0.001 6 mmol of catalyst. After being stirred at room temperature for 24 h, the mixture was treated with saturated ammonium chloride solution and extracted with ethyl acetate. The organic layer was dried (MgSO4), filtered and concentrated to give Aldol product after thin layer chromatographic purification on silica gel (petroleum ether/ethyl acetate).
4-hydroxy-4-(2-nitrophenyl)butan-2-one1H NMR (CDCl3)δ: 2.24 (s, 3H), 2.74 (dd,J= 17.8, 9.4 Hz, 1H), 3.13 (dd,J= 17.8, 1.8 Hz, 1H), 3.89 (s, 1H), 5.68 (dd,J= 9.4, 1.8 Hz, 1H), 7.44 (t,J= 8.0 Hz, 1H), 7.67 (t,J= 8.0 Hz, 1H), 7.80 (d,J= 8.0 Hz, 1H), 7.96 (d,J= 8.0 Hz 1H); HPLC: Chiralcel OD-H, UV 254,i-PrOH/Hexane = 5/95, 1.0mL/min,S-isomer,tR18 min,R-isomer,tR21 min.
2-(Hydroxyl(4-nitrophenyl)methyl)cyclohexanone1H NMR (CDCl3)δ:syn-isomer: 1.50-1.88 (m, 5H), 2.09-2.15 (m, 1H), 2.37-2.52 (m, 2H), 2.62-2.66 (m, 1H), 3.10 (s, 1H), 5.49 (d,J= 1.6 Hz, 1H), 7.49 (d,J= 8.4 Hz, 2H), 8.22 (d,J= 8.4 Hz, 2H);anti-isomer: 1.36-1.44 (m, 1H), 1.51-1.73 (m, 3H), 1.83 (m, 1H), 2.10-2.15 (m, 1H), 2.33-2.46 (m, 1H), 2.50 (m, 1H), 2.57-2.63 (m, 1H), 3.80 (s, 1H), 4.90 (d,J= 8.4 Hz, 1H), 7.51 (d,J= 8.4 Hz, 2H), 7.22 (d,J= 8.4 Hz, 2H); HPLC (foranti-isomer): Chiralcel OD-H, UV 254,i-PrOH/Hexane = 5/95, 1.0 mL/min,tR29 min (major),tR45 min (minor).
2-(Hydroxyl(2-nitrophenyl)methyl)cyclohexanone1H NMR (CDCl3)δ:syn-isomer: 1.53-1.87 (m, 5H), 2.10 (m, 1H), 2.42-2.47 (m, 2H), 2.90 (dd,J= 13.2, 4.8 Hz, 1H), 3.15 (s, 1H), 5.96 (d,J= 1.6 Hz, 1H), 7.46 (dt,J= 0.8, 8.0 Hz, 1H), 7.66 (dt,J= 0.8, 8.0 Hz, 1H), 7.84 (dd,J= 8.0, 0.8 Hz, 1H), 8.02 (dd,J= 8.0, 0.8 Hz, 1H);anti-isomer: 1.61-1.87 (m, 5H), 2.10 (m, 1H), 2.34-2.47 (m, 2H), 2.77 (m, 1H), 3.95 (s, 1H), 5.45 (d,J= 7.2 Hz, 1H), 7.44 (dt,J= 0.8, 8.0 Hz, 1H), 7.66 (dt,J= 0.8, 8.0 Hz, 1H), 7.78 (dd,J= 8.0, 0.8 Hz, 1H), 7.86 (dd,J= 8.0, 0.8 Hz, 1H); HPLC (foranti-isomer): Chiralcel OD-H, UV 254,i-PrOH/Hexane = 5/95, 1.0 mL/min,tR17 min (major),tR21 min (minor).
2-(Hydroxyl(4-chlorophenyl)methyl)cyclohexanone1H NMR (CDCl3)δ:syn-isomer: 1.42-2.11 (m, 6H), 2.32-2.45 (m, 2H), 2.53-2.56 (m, 1H), 3.05 (s, 1H), 5.36 (d,J= 2.0 Hz, 1H), 7.24 (d,J= 8.4 Hz, 2H), 7.32 (d,J= 8.4 Hz, 2H);anti-isomer: 1.27-1.31 (m, 1H), 1.53-1.82 (m, 4H), 2.07-2.11 (m, 1H), 2.35-2.56 (m, 3H), 4.76 (d,J= 8.8 Hz, 1H), 7.26 (d,J= 8.0 Hz, 2H), 7.32 (d,J= 8.0 Hz, 2H); HPLC (foranti-isomer): Chiralcel OD-H, UV 220,i-PrOH/Hexane = 5/95, 1.0 mL/min,tR14 min (major),tR22 min (minor).
2-(Hydroxyl(3-nitrophenyl)methyl)cyclohexanone1H NMR (CDCl3)δ:syn-isomer: 1.48-2.10 (m, 6H), 2.33-2.48 (m, 2H), 2.62-2.66 (m, 1H), 3.16 (s, 1H), 5.48 (d,J= 2.0 Hz, 1H), 7.52 (t,J= 8.0 Hz, 1H), 7.66 ( d,J= 1.4 Hz, 1H), 8.11 (d,J= 8.0 Hz, 1H), 8.20 (d,J= 8.0 Hz, 1H);anti-isomer: 1.33-2.10 (m, 6H), 2.32-2.48 (m, 2H, ), 2.70 (m, 1H), 3.16 (s, 1H), 4.91 (d, J = 8.4 Hz, 1H), 7.54 (t,J= 8.0 Hz, 1H), 7.68 (d,J= 0.8 Hz, 1H), 8.15 (m,J= 8.0 Hz, 1H), 8.20 (d,J= 8.0 Hz, 1H); HPLC (foranti-isomer): Chiralcel OD-H, UV 254,i-PrOH/Hexane = 5/95, 1.0 mL/min,tR24 min (major),tR36 min (minor).
2Results and Discussion
2.1Synthesis of the catalysts 1a-b
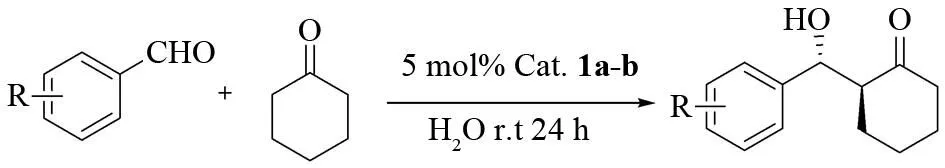
Two diastereomers 4-sulfonamido-L-prolines were synthesized easily from the commercially availabletrans-4-hydroxy-L-proline with overall yields of 55% and 70% respectively (Fig.1). The spatial configuration was controlled under different reaction conditions when N-Cbz-4-hydroxy -L-proline methyl ester reacted with 4-tosyl chloride. Compound 3a may be generated by two steps of nucleophilic substitution reactions (Fig.2). The structures of the two catalysts were characterized by IR,1H NMR and HR-MS spectra.
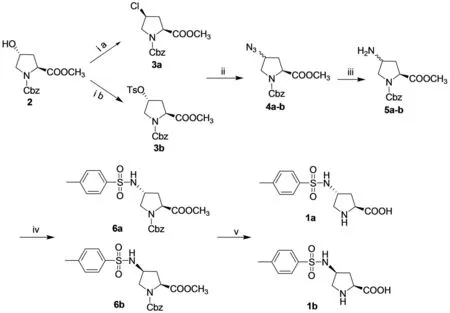
Reagents and conditions: (ia) TsCl, pyridine, 70℃; (ib) TsCl, pyridine, r.t; (ii) NaN3, DMF, 65℃;(iii) PPh3, THF, reflux; (iv) TsCl, Et3N, CH2Cl2, r.t; (v) ① 10% NaOH aq./THF; r.t ② H2, Pd/C, EtOH.Fig.1 Synthesis of the catalysts 1a-b
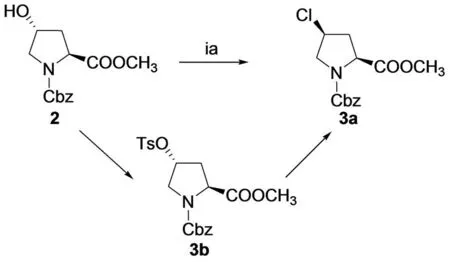
Fig.2 Probable reaction mechanism
2.2Aldol reactions
To evaluate the catalytic efficiency of the two diastereomeric catalysts 1a-b, the reaction ofo-NO2benzaldehyde with acetone was first performed. The results, as listed in Table 1, indicated that the catalysts 1a and 1b have the similar catalytic efficiency. The yields of the Aldol reactions are moderate to good, however, theeevalue are comparable to other small moleculeL-proline derivatives (Entries 1-2). It has been shown that water could play a special role in the catalyzed Aldol reaction in aqueous (homogeneous) media. The direct Aldol reaction in water/aqueous media has been developed in recent years. We also tested the catalytic effect of 1a-b in ketone/water mixture (Entries 3-14). To our disappointed, with the increase of the water quantity, the yield decreased gradually accompany a sharp decrease in the enantioselectivity. At the same time, the absolute configuration of product has been reversed. However, when equal molar amount of zinc chloride was added, the structure of the product will be reversed again (Entries 15-18).
Table 1Direct Aldol reaction of acetone witho-NO2benzaldehyde catalyzed by 1a-b at room temperature
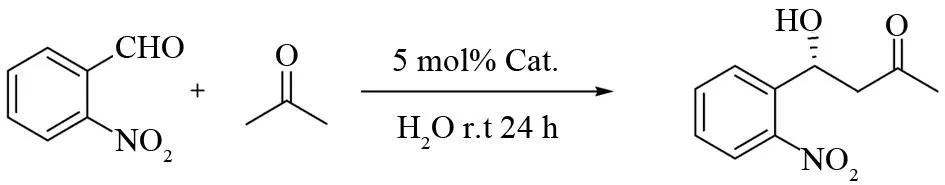
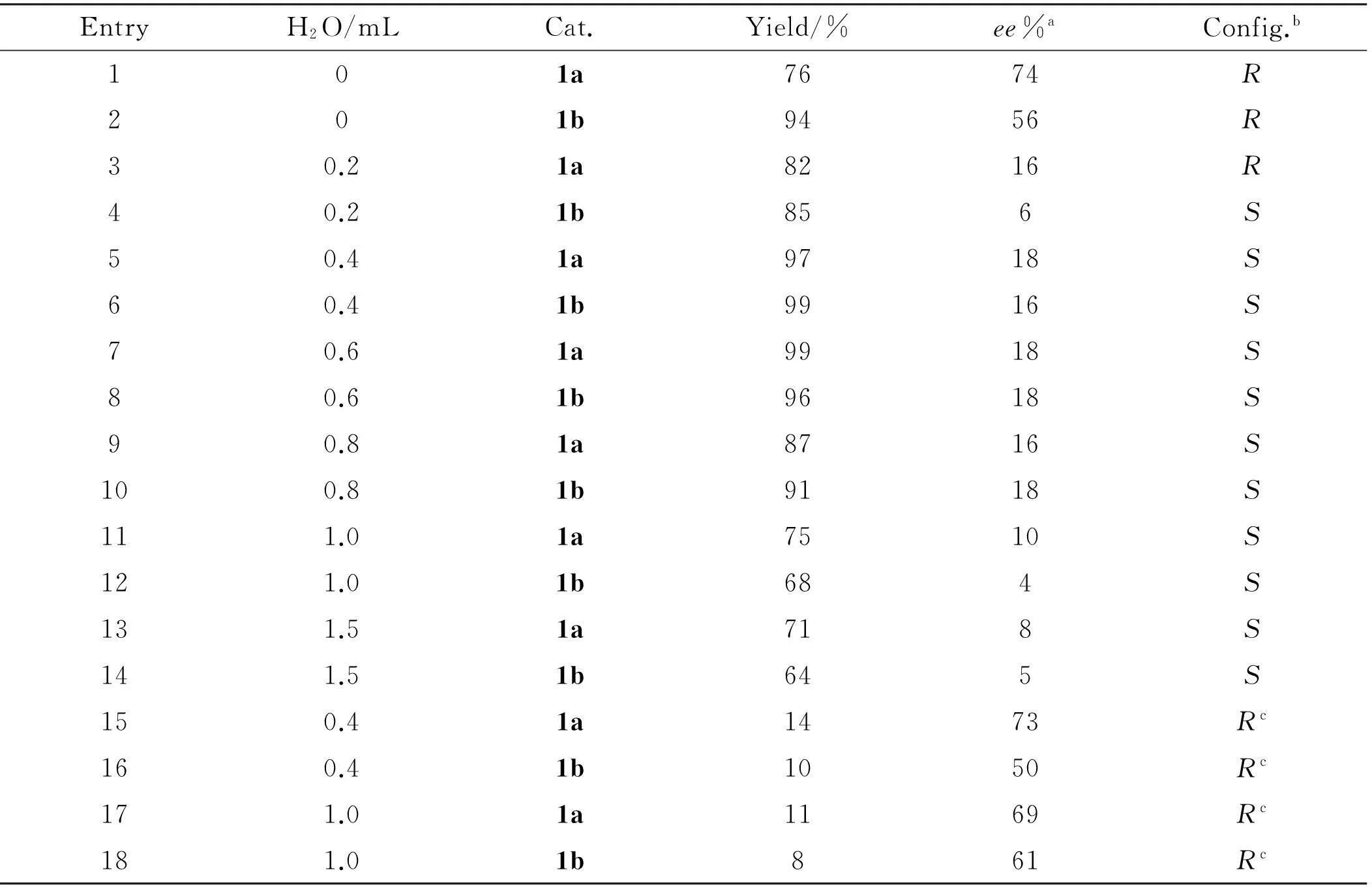
EntrypO/mLCat.Yield/%ee%aConfig.b101a7674R201b9456R30.21a8216R40.21b856S50.41a9718S60.41b9916S70.61a9918S80.61b9618S90.81a8716S100.81b9118S111.01a7510S121.01b684S131.51a718S141.51b645S150.41a1473Rc160.41b1050Rc171.01a1169Rc181.01b861Rc
aDetermined by chiral HPLC on a chiralcel OD-H column.
bAccording to reference
cEqual molar amount of zinc chloride was added
In order to check the versatility of the catalysts 1a-b, the reaction between cyclohexanone and several other substituted benzaldehydes were investigated in water at room temperature. The results, as listed in Table 2, indicated that the yields of the Aldol reactions are good to excellent. The results also showed that catalysts 1a and 1b have the similar catalytic efficiency.p-Chlorobenzaldehyde showed lower activity than nitrobenzaldehyde, however, the diastereoselectivity and theeevalues are comparable.
Table 2Direct Aldol reaction of cyclohexanone with aldehyde catalyzed by 1a-b in water at room temperature
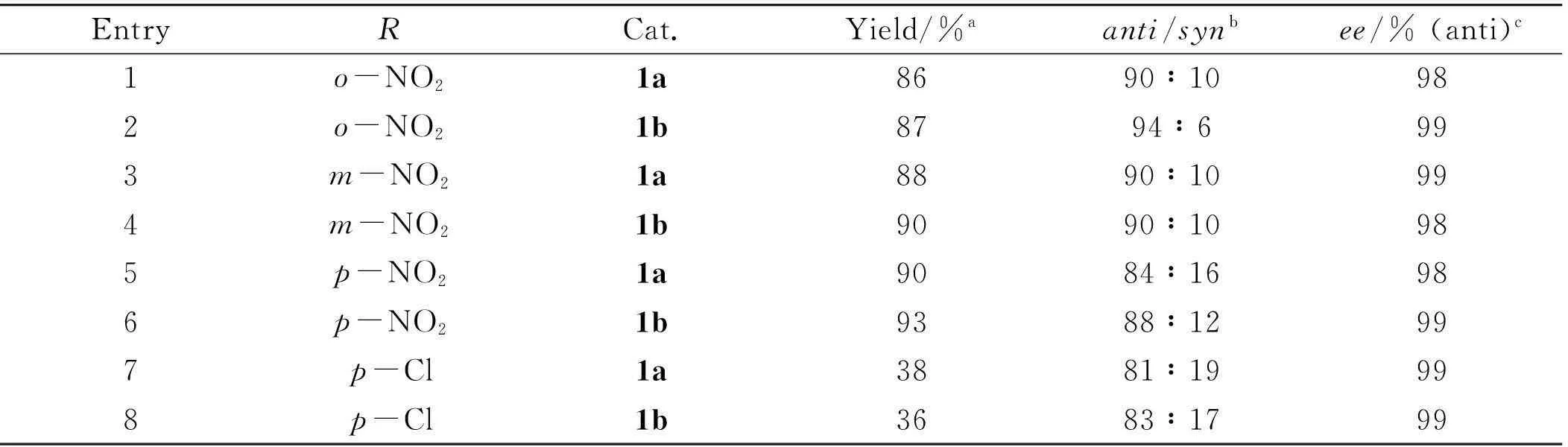
EntryRCat.Yield/%aanti/synbee/%(anti)c1o-NO21a8690∶10982o-NO21b8794∶6993m-NO21a8890∶10994m-NO21b9090∶10985p-NO21a9084∶16986p-NO21b9388∶12997p-Cl1a3881∶19998p-Cl1b3683∶1799
aIsolated yields (anti+syn) after thin layer chromatography on silica gel.
bDetermined by analysis of1H NMR spectra of the mixture ofantiandsynproducts.
cDetermined by chiral HPLC on a chiralcel OD-H column.
3Conclusions
In conclusion, two diastereomers 4-sulfonamido-L-prolines 1a-b have been synthesized for asymmetric direct Aldol reaction. The catalytic effect of these catalysts for the Aldol reaction was evaluated in water. Good yields and high stereoselectivity (93% yield, 94∶6anti/synratio, 99%ee) were obtained for the reactions of aromatic aldehydes with ketones. It was also found that water could play a special role to improve the reactivity and stereoselectivity in the Aldol reaction catalyzed by 4-sulfonamido-L-prolines 1a-b.The research expand the scope of organic catalysts in the field of enantioselective synthesis, making the search for these reagents more appealing.
References:
[2] SUTAR R L, JOSHI N N. Systematic evaluation of a few proline derivatives as catalysts for a direct Aldol reaction [J]. Tetrahedron: Asymmetry, 2013, 24: 43-49.
[4] LIU Y X, SUN Y N, TAN H H, et al. Asymmetric Aldol reaction catalyzed by new recyclable polystyrene-supported L-proline in the presence of water [J]. Cat Lett, 2008, 120: 281-287.
[5] LIU Y X, SUN Y N, TAN H H, et al. Linear polystyrene anchored L-proline, new recyclable organocatalysts for the Aldol reaction in the presence of wate [J]. Tetrahedron: Asymmetry, 2007, 18: 2649-2656.
[6] PELLISSIER H. Recent developments in asymmetric organocatalytic domino reactions [J]. Adv Synth Catal, 2012, 354: 237-294.
[8] MOLLETI N, RANA N K, SINGH V K. Highly enantioselective conjugate addition of malononitrile to 2-enoylpyridines with bifunctional organocatalyst [J]. Org Lett, 2012, 14: 4322-4325.
[9] POWELL A B, SUZUKI Y, UEDA M, et al. A recyclable, self-supported organocatalyst based on a poly(N-heterocyclic carbene) [J]. J Am Chem Soc, 2011, 133: 5218-5220.
[10] BHANJA C, JENA S, NAYAK S, et al. Organocatalytic tandem Michael addition reactions: A powerful access to the enantioselective synthesis of functionalized chromenes, thiochromenes and 1,2-dihydroquinolines [J]. Beilstein J Org Chem, 2012, 8: 1668-1694.
[12] AN Y J, ZHANG Y X, WU Y, et al. Simple amphiphilic isosteviol-proline conjugates as chiral catalysts for the direct asymmetric Aldol reaction in the presence of water [J]. Tetrahedron: Asymmetry, 2010, 21: 688-694.
[13] GRUTTADAURIA M, RIELA S, APRILE C, et al. Supported ionic liquids. new recyclable materials for theL-proline-catalyzed Aldol reaction [J]. Adv Synth Cata, 2006, 348: 82-92.
[14] HAYASHI Y, ARATAKE S, ITOH T. Dry and wet prolines for asymmetric organic solvent-free aldehyde-aldehyde and aldehyde-ketone Aldol reactions [J]. Chem Commun, 2007: 957-959.
[15] ZHOU J Q, WAN J W, MA X B, et al. Copolymer-supported heterogeneous organocatalyst for asymmetric Aldol addition in aqueous medium [J]. Org Biomol Chem, 2012, 10: 4179-4185.
[责任编辑:任铁钢]
