
铑基催化剂用于低碳醇合成反应研究进展
2016-05-17 08:42韩通赵琳岳义智刘源天津大学化工学院天津300072
化工进展 2016年4期
韩通,赵琳,岳义智,刘源(天津大学化工学院,天津 300072)
铑基催化剂用于低碳醇合成反应研究进展
韩通,赵琳,岳义智,刘源
(天津大学化工学院,天津 300072)
摘要:分别从反应机理、活性位点、助剂和载体4个方面对合成气制低碳醇铑基催化剂体系进行了文献综述,同时对理想催化剂的研究方向做出了预测。通过对不同学者的研究成果的对比,发现对于铑基催化剂催化的反应机理和活性中心等方面的研究仍存在较多争议,更为深入仔细的研究工作仍然需要。应用前景方面,虽然铑基催化剂对该反应具有较高的低碳醇选择性(尤其是乙醇)和较好的稳定性,但较低的反应活性及铑金属自身昂贵的价格很大程度上限制了其应用,具有工业应用价值的高效铑基催化剂仍有待开发。
关键词:合成气;催化剂;醇;载体;稳定性
第一作者:韩通(1990—),男,硕士研究生。联系人:刘源,教授,博士生导师,主要从事双金属催化剂用于低碳醇合成反应的研究。E-mail yuanliu@tju.edu.cn。
随着能源危机的加剧和环境的恶化,新型环保能源的研发显得越发重要。低碳醇作为一种优良的汽油添加剂,不仅可以提高汽油的辛烷值,还可以减少来源于传统化石燃料燃烧产生的SO2等污染气体的排放,近年来引起了人们的广泛关注。由煤炭合成气(CO+H2)合成低碳混合醇是一条可行的技术路线,自20世纪中后期被提出以来,一直都是国内外的研究热点。
合成气可以由固体煤燃料气化制得,由合成气直接转化成以乙醇为主的低碳混合醇,一方面可以在一定程度上改善我国富煤少油的尴尬现状,减轻石油资源枯竭带来的危害;另一方面,同样可以减少我国的粮食消耗,减轻粮食危机,对于我国的可持续发展具有重要的意义。
产物种类分布不集中、C2+醇选择性差、CO转化率较低是困扰低碳醇合成的主要问题。因此制备具有高活性、高C2+醇选择性且稳定性良好的催化剂是低碳醇合成的核心问题,也是国内外众多催化研究者们研究的热点。1975年,美国联碳公司报道了一种用金属铑作活性组分的负载型催化剂[1],可通过CO加氢反应得到高产率的C2+醇和醛等产物。
从那时起,铑基催化剂吸引了众多研究者的兴趣。一直以来,铑基催化剂以其较高的醇类(特别是乙醇)选择性备受国内外学者的青睐。研究者们的研究重点主要放在了以下几个方面:反应机理的探究,催化活性中心位的研究,合适助剂、载体的寻找以及反应活性与选择性的提高。本文主要对近年来报道的铑基催化剂用于低碳醇合成的文献进行了综述,从以上几个方面对该类催化剂的研究现状进行总结和归纳,并进行展望。
1 铑基催化剂上C2+氧化物的生成机理
无论使用何种催化剂,合成气加氢反应的产物种类分布都较为广泛,不仅有甲醇、甲烷、H2O等小分子物质,还有较长链的烯烃、烷烃、醛、羧酸以及目标产物C2+醇。张永青等[2]认为这种产物分布符合Schuiz-Flory分布,这种产物分布同样符合目前广泛被人们接受的CO解离-插入机理。
众所周知,产物种类的复杂性必然由其复杂的反应机理决定。而研究一种催化剂关键就是了解其催化的反应机理,从而控制产物的分布。因此,在铑基催化剂的研究初期,众多研究者的工作重点都放在解释低碳醇合成反应的复杂的反应机理上。对于铑基催化剂的催化反应机理,在那段时期各种观点层出不穷,具有代表性的有以下几种。
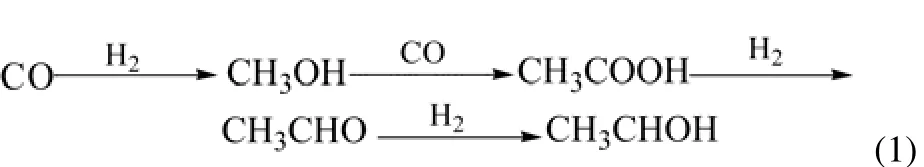
(1)甲醇同化类 持有这类观点的学者认为:甲醇是乙醇的前驱物种,CO加氢主要生成甲醇,生成的甲醇和剩余的CO进一步羰基化生成乙酸,生成的乙酸可以进一步加氢生成乙醛,乙醛仍可以进一步加氢生成目标产物乙醇以及更长链的醇[3],如式(1)。
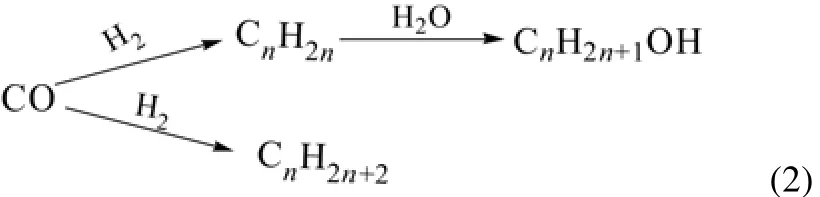
(2)烯烃水合系 这类机理中,参与反应的CO均发生解离吸附,生成的碳物种和氢气发生反应生成烃类(烷烃、烯烃),其中的烯烃可以和水蒸气进一步反应生成低碳混合醇[4],如式(2)。
(3)CO解离-插入机理 TAKEUCHI等[7]通过多年的研究提出合成气制备C2+含氧化合物的详细插入模式,如图1。一部分CO在催化剂活性位表面发生解离吸附,吸附的碳物种与氢气反应,生成表面活性烷基物种CHx,CHx物种可以继续加氢生成烃类物种,该物种脱附即可得到CH4及更长链的烷烃和烯烃;CHx物种还可以和另一部分未解离吸附的CO发生反应,通过插入过程进入到烷基物种,生成含氧的不饱和中间物,该中间物进一步加氢并脱附得到含氧化合物(甲醇、目标产物C2+醇、醛及羧酸类物质)。

图1 CO解离-插入机理[5-10]
对比3种铑基催化剂上的反应机理不难发现:第一种机理只对甲醇和乙醇的生成做了解释,对于副产物烃及其他含氧化合物的存在并没有给出合理的解释,故这种机理不符合实际产物分布,不具备说服力;第二种机理认为一氧化碳只能氢助解离生成烃类,且醇类的生成需要水蒸气参与反应,可以明显发现该机理不能解释甲烷、甲醇等主要副产物的生成且反应物中并没有大量的水蒸气存在,因此,可以同样认为该机理不具备说服力。而第三种机理无论是对低碳混合醇等目标产物还是甲烷、甲醇、烃类等主要副产物都做出了合理的解释。因此在该研究领域这种机理(CO解离-插入机理)被普遍接受。众多学者对该机理给出了解释,最具有说服的是ORITA和ICHIKAWA等[9-10]的同位素实验:他们首先通过CO歧化反应在催化剂表面铺上一层标记的13C,然后在反应条件下和12C/H2混合气接触反应发现,13CH3CHO、13CH3H2OH的瞬时浓度很高,13C在醛基上和在羟基碳原子上的乙醛和乙醇浓度很低。这说明乙醛、乙醇等含氧化合物的生成是通过未解离吸附的CO插入表面碳物种而来。而且,ORITA等[9]的进一步同位素示踪法表明:乙醇中的CH3和烃类产物中的CHx(x为2或3)来自共同的C1物种CHx。这就是说在生成一个乙醇分子中的两个CO分子中,其中一个在发生链增长反应(如CO插入)之前必须断裂C—O键。
第三种机理(CO解离-嵌入机理)的提出基本上结束了研究者对于CO活化、参与反应方式的争论。此后的文献中,研究者几乎都是以该机理为基础进行探究[11-13]。近年来,随着表征手段的多样化以及表征精度的提高,有关这方面的文献主要集中在对该机理中某一具体步骤详细解释或修正以及目标产物前体(反应关键活性物种)的判别等方面。
MAO[14]、LIU[15]、YU[16]等认为,在CO解离之前应首先与氢气反应形成CH2OH*活性物种,该物种发生解离生成CH2*活性物种,CH2*发生上述类似的链增长与非解离CO嵌入反应生成相应产物。CH2OH*发生解离的同时会伴随大量OH*物种的生成,该物种与H*及CH2CO*物种生成H2O与CH3COOH等副产物。该解释完善了这类机理关于CO具体解离方式的阐述,同时更好地阐述了H2O、CH3COOH等副产物的生成机理。
CHx(x=1~3)种类不一,活泼性不同,其中最活泼、最利于接受非解离CO嵌入的表面CHx物种对反应意义巨大。寻找这类物种同样是近年的热点:CHOI等[17]认为Rh(111)表面吸附的CH3*物种最为活泼,最利于接受CO*,对C2+含氧化合物生成起主导作用;C2+含氧化合物的选择性主要取决于CH3*物种加氢反应与CO嵌入反应的相对反应位垒的相对大小。KAPUR等[18]则给出了完全相反的结论,他们认为CH2*物种对Rh(111)表面C2+含氧化合物的生成起主导作用,CO嵌入CH2*物种形成的C—C中间体是含氧化物的主要前体。
GAO等[19]对Rh/ZrO2催化剂进行了基于反应活性位水平上的SSITKA实验得出以下结论:甲烷与甲醇在完全不同的活性位上生成;乙烷(乙烯、乙炔)、乙醇不是由乙酸基物种进一步反应生成;所有的C2产物均与甲烷都至少有一个相同的活性基团构成,即烷基物种对C2+含氧化合物选择性作用巨大,甲醇类活性物种对其生成没有影响。
2 活性中心原子价态
除关于对反应机理的争论外,关于催化剂活性中心铑纳米颗粒的价态问题同样存在着不小的争议,铑基催化剂的争议主要集中在+1价态的铑离子(Rh+)是否存在的问题上。早期文献中,较多的学者认为催化活性中心中必须含有铑离子,且铑离子起到重要的催化效果:WASTON和SOMORJAI[20-21]等发现,只有含Rh+离子的催化剂才有C2+含氧化合物生成,他们提出一个双功能催化机理:H2、CO的解离发生在还原态的Rh上,生成CHx中间体,CO的插入过程则发生在Rh+离子上;他们认为Rh和Rh+都是生成C2+含氧化物的活性位。包信和等[22]在近几年的文献中也指出,提高Rh+/Rh的比值有利于提高插入的CO原子比例,可以增加C2+含氧化物的生成量;DUSHYANT等[23]通过对比了金属铑进入黄绿矿(La2Zr2O7,LZ)晶格催化剂和金属铑负载在黄绿矿上的催化剂的不同性能,得出了稳定催化剂中的Rh+的存在有利于减少二氧化碳的生成,同时提高C2+含氧化合物的选择性的结论;VAN DEN BENG等[24]用化学提取法发现,在高活性的Rh-Mn-Mo/SiO2催化剂中有大量的Rh+,而活性较低的Rh/SiO2中Rh+较少;这些结果都有力支持了Rh+为活性中心的观点。
近年来,不少学者却提出了相反的论断,他们认为铑基催化剂的活性中心没有铑离子的存在,所有的催化反应均是在铑原子表面进行的。KAZTER 等[25]报道了XPS的研究结果,发现在Rh/TiO2上没有明显的证据证明Rh+物种的存在,但该催化剂却对合成C2+含氧化合物有活性。陈明英等[26]对Rh-Mn/SiO2催化剂上CO的吸附进行了红外光谱研究,发现在 CO 气氛中,当温度升至 200℃时,孪生 CO 吸附物种消失,表明在反应温度下可能不存在 Rh+物种,即 Rh+物种可能不是 CO 插入的活性位。
由此可见,要弄清楚到底单单是Rh原子还是Rh+/Rh双功能作用对合成C2+含氧化合物有贡献仍有待进一步的实验研究。但无论支持哪种观点的学者都一致认为:反应活性和选择性对活性金属粒度的大小和负载量的变化都是非常敏感的;合适的活性中心纳米颗粒的粒度和负载量是高活性和选择性的保障。
HANAOKA等[27]对粒径1~6nm的铑纳米颗粒的活性基团进行了CO加氢实验,发现只有粒径大于等于2.5nm的铑纳米颗粒对于该反应才有较高的活性和选择性,当粒径过小时,反应产物主要为甲醇。MANUEL等[28]制备了不同粒径的铑纳米颗粒,对比不同催化剂的性能发现:较大的铑纳米颗粒有助于提高催化剂反应活性和选择性,有助于CO的解离吸附,但烃类产物较多。针对这种现象,作者并没有确切的解释,但是给出了两种合理的假设:①催化剂铑纳米颗粒表面晶面缺陷是CO解离的主要活性位,粒径较大的铑纳米颗粒具有较多的表面阶跃和缺陷,因此具有较高的解离活性;②CO解离生成的活性碳和原子分别吸附在相邻的两个铑原子上,且链增长生成较长链的CHx物种也需要较大的铑纳米颗粒支撑,故粒径大的催化剂具有较高的解离活性;另一方面,较小的铑纳米颗粒有助于CO的非解离吸附,此时催化剂具有较高的含氧化合物选择性,但反应活性较低。
关于铑的负载量,文献主要集中在最佳负载量的寻找上:ICHIKAWA[29]研究了分散度对Rh/SiO2催化剂性能的影响,他们的研究工作表明C2+含氧化物在Rh分散度约为0.40(Rh担载量质量分数4.7%)时达到最大值。尹红梅[30]的最佳Rh负载量优选实验中得到如下结论:C2含氧化物的选择性在Rh担载量约为1%~2%(质量分数)时达到最大值,相应Rh的分散度约为0.35~0.51,两者的结论基本吻合。
3 铑基催化剂助剂研究状况
由于未负载或者惰性载体(本身无催化活性)负载的铑基催化剂对该反应的反应活性和选择性都比较低,且产物以甲烷为主。因此在该体系的研究中,学者们最主要的工作是寻找合适的助剂并探究其作用机理,以期获得良好的反应活性和选择性。助剂的种类繁多,不仅有过渡金属还包含部分碱金属和贵金属,且不同种类的助剂作用效果也有显著的区别。讲述单一助剂作用的催化剂都是较早的文献,近期的文献中报道的工作主要集中在以下3个方面。
3.1助剂作用综述及分类
CHUANG等[31]的外文综述中将助剂按照其作用进行了分类:ZnO、MgO等碱金属有助于CO加氢形成甲醇;Mn、V、Na、La等金属有助于CO的解离;Fe、Ir、P等有助于C物种在催化剂活性表面加氢;Ag、Zn、La、V有助于CO的插入反应。GONZALO等[32]对低碳醇合成铑基催化剂氧化物助剂电子性质的作用进行了新的展望。他们认为,无论是催化剂的活性还是选择性,都跟助剂的电子性质密切相关。不同电子类型的催化剂作用不同。
吸电子型助剂(Ta、Y、V、Ti、Nb)的添加使得催化剂具有较高的加氢活性和烃类选择性,作者给出的解释是该种助剂的添加使得桥式吸附的CO(Rh2COB)和原子态的Rh0表面电子云密度增加,这两种情况都会造成CO的解离吸附增加;供电子型助剂(Sm、Nd、Pr、Fe)的添加使得催化剂具有较好的醇类选择性,作者给出的解释主要是该类型助剂使得线式吸附的CO对活性铑原子具有更少的电子反馈,使得CO的非解离吸附增加,从而增加氧化物的选择性。
3.2合适助剂组合的寻找
MO等[33]分别制备了V、La、Fe单独负载、La-V、La-Fe以及La-Fe-V共同负载的铑基催化剂,通过对比发现:V助剂的添加能够增加CO的脱附速率和CO在催化剂表面的反应活性;La助剂的添加可使催化剂表面产生新的吸附物种,而这类吸附物种的出现提高了反应活性;Fe助剂的添加显著降低了烃类选择性,但甲烷和甲醇的选择性较高;La-V修饰的催化剂比单金属修饰的催化剂具有更多吸附CO的活性位,活性更高,但选择性较低;La-Fe系催化剂具有较高的活性和乙醇选择性,但甲烷的选择性同样较高;La-Fe-V三金属负载的催化剂在表现出较好的反应活性和选择性的同时具有较低的甲烷和甲醇等副产物的选择性。大连化学物理研究所的尹红梅[30]报道的Mn-Li-Fe催化剂较单金属或双金属负载的催化剂同样具有较好的反应活性、较高的低碳醇选择性以及较低的副产物选择性,大连化学物理研究所还对该催化剂进行了中试,表现出了良好的应用前景。
3.3理论计算进一步阐述助剂的作用
LIU等[34]对过渡金属掺杂的铑基催化剂进行了密度泛函动力学蒙特卡罗模拟研究得出如下结论:金属掺杂和掺杂金属的位置影响低碳醇合成反应的活性和选择性,合金催化剂制备过程中控制掺杂金属的位置对催化剂活性至关重要。具体表现在:对于掺杂的过渡金属Mo和Mn,不同的掺杂位置具有不同的反应活性和乙醇选择性,分布在铑表面的Mo掺杂催化剂活性比位于次表面的催化剂要高,而对于Mn金属来说情况恰好相反,位于次表面层的Mn具有更高的反应活性和乙醇选择性。ZHAO 等[35]以密度泛函理论为基础,对该体系进行了半定量的动力学分析发现:Cu(111)修饰的Rh-Cu(111)双金属合金较单独的Rh(111)或附近的Rh(553)具有较高的醇类选择性和较低的甲烷选择性,且铜掺杂的催化剂具有极强的抗烧结性能和较低的价格,作者因此得出结论:铜作为提高反应醇类选择性的催化剂替代材料是非常有前景的。
除了以上3个方面,助剂的作用机理也是学者们关注的热点,虽然没有文献专门介绍,但每篇文献都会有涉及这方面的报道。通过对不同的文章进行分析比较,研究者们的观点基本可以统一为:助剂主要通过改变CO在活性位Rh表面的吸附强度和吸附物种来改变催化剂的活性[11]:La、Mn助剂[33]的文献中都认为金属和助剂表面倾斜吸附的CO是催化剂活性提高的主要原因,这种吸附可以降低CO中碳氧键强度,使其更有利于解离从而增加反应活性。陈明英等[26]还发现:Mn 的添加还有助于削弱线式 CO 吸附物种的 Rh–C键,使其更易于在表面上迁移,进而有利于CO的插入,提高C2含氧化合物的选择性。MO等[33]文献中也提到线式和倾斜吸附CO的增加有利于反应活性和选择性的提高,不过文献中并未给出合理的解释。
4 催化剂载体的研究状况
如同助剂一样,载体的选择同样可以改变催化剂的性能。研究者们发现铑基催化剂的活性组分铑与载体之间存在着很强烈的相互作用,这种相互作用被称为SIMI效应,最早由TAUSTER等[36]报道。根据文献内容大致可以将载体分为以下几类。
4.1惰性载体
惰性载体单纯的起负载活性组分的作用,对活性组分的活性没有影响。早期文献认为,这类载体主要包括Al2O3和SiO2两种,在文献中主要用作空白对照以突出某种催化剂的某种特殊性能。MANUEL等[28]采用溶胶-凝胶法制备了Al2O3负载的具备不同粒径的铑基催化剂,通过对比催化剂活性得出了结论,并将活性差异完全由归咎于铑纳米颗粒大小的差异,认为载体Al2O3无任何活性。GONZALO等[32]探究不同类型助剂作用的文献也是通过将活性组分和助剂分散在Al2O3表面进行研究的。SiO2较Al2O3应用更为广泛,较早的研究者认为在SiO2担载的Rh基催化剂用于C2含氧化合物的合成反应中,载体SiO2是没有活性的,对反应没贡献,属于惰性载体。但近年的研究工作给出了相反的结论,下文将做具体叙述。
4.2活性载体
活性载体是除了负载活性组分的作用外,还对催化剂的活性组分起到修饰作用,从而改善反应活性或选择性的载体。这类载体的种类较多,应用较广泛。其中活性较好的主要有SiO2、TiO2、ZrO2等。早期SiO2一直被认为是惰性载体,而一些研究工作表明,金属和助剂等组分可能是通过与SiO2表面的羟基相互作用而锚合在载体上的[37],从而制得高度分散的催化剂,这一发现使得学者不再认为SiO2是简单的惰性载体。于军等[38]采用Stöber方法制备了一种新型SiO2作为载体,发现该催化剂具有较好的活性和选择性。他们认为主要是这种新型的SiO2载体表面羟基与金属间的相互作用改变了CO在金属铑纳米颗粒表面的吸附,从而影响了CO的加氢活性。IOANNIDES等[39]发现TiO2负载的铑基催化剂相比SiO2负载催化剂具有更好的活性和选择性,并将这种差别归因于TiO2和SiO2不同的表面和纹理结构。ICHIKIWA[40]制备的ZrO2负载的铑基催化剂获得了接近60%的乙醇选择性,表明氧化锆载体对铑基催化剂有着特殊的修饰促进作用。
4.3复合载体
复合载体是指将两种或多种载体通过某种方法共同制备,以期达到单一载体不具有的催化活性和选择性。由于SiO2的特殊性质,文献中基本都是将氧化硅与另一类载体复合进行研究的。HAN等[41]制备了TiO2-SiO2复合载体,发现相比于单一类型的TiO2或SiO2载体,该复合载体对该体系的活性组分Rh具有较高的分散度,使得该催化剂具有较多的Rh+以及中等程度的加氢能力,从而具有更好的反应活性和选择性。他发表的另一篇文献[42]中,采用浸渍法分别制备了Si/Zr比为1∶1、1∶3、1∶9的SiO2-ZrO2复合载体,发现Si/Zr比为1:3的复合载体表现出最好的反应活性和醇类选择性,这些性能同样是单一载体负载的催化剂所不具有的。
4.4新型碳材料载体
相比于传统载体,碳材料载体具有与活性组分更好或者某种特殊的相互作用,从而展现出新的应用前景。FAN等[43]重点研究了碳材料载体的性能差别,分别制备了以CNT、炭黑、CMK-3分子筛为载体的催化剂,发现在众多载体中CNT负载的催化剂具有最好反应活性及选择性;他们还对具有类似孔道结构的CNT和CMK-3分子筛进行了专门对比,发现虽然具有类似的孔道结构和孔径,CNT具有更好的活性,从而突出了CNT作为载体的优越性。作者认为该种现象是CNT和金属纳米粒子之间独特的强烈相互作用造成的。MAO等[14]分别制备了CNT外壁和内部孔道负载的铑基催化剂,发现内部孔道负载的铑基催化剂比外壁负载的铑基催化剂活性要高出好几个数量级,他认为碳纳米管内部孔道的缺电子性能促进CO和铑纳米颗粒间的作用,从而产生了高活性和选择性。近年来,CNT作为载体或促进剂的研究报道层出不穷。
5 催化剂制备方法
浸渍法是该体系催化剂的主要制备方法,无论是讲述反应机理还是助剂、载体作用的文章基本都是采用这种方法制备的,这种相对简单的制备方法也使该体系催化剂的工业应用变成了可能。MO课题组[33]的La-V-Fe体系催化剂表现出了良好的活性和选择性,就是采用浸渍法制备的。值得注意的是,在文献中作者根据不同物质性质的不同采用的浸渍顺序是不同的:La、Fe的硝酸盐可以共存,但钒酸盐难溶且不共存,故采用La、Fe共同浸渍,之后再浸渍V的方法制备。除浸渍法之外,为达到特定的目的,文献中也有采用其他方法制备的,如MANUEL Ojeda等[28]探究载体表面纳米颗粒大小对催化剂活性影响的文献中,为了得到不同粒径的铑基催化剂,采用的就是溶胶-凝胶的方法对催化剂进行了制备。XU等[44]探究的分子筛载体作用文献则采用的是以分子筛为基础的离子交换法制备的催化剂。
虽然研究者对这一体系催化剂不同组成部分均进行了详尽的研究。但无论该催化剂活性中心铑粒子具有何种形状、价态或者使用何种载体、助剂,CO在活性铑粒子表面的吸附类型及活化方式都是反应活性的决定因素,不同吸附物种间的竞争以及目标吸附物种生成及脱附速率则是醇类选择性高低的决定因素。
不同作用的助剂对铑粒子的吸附调变作用不同:La、Mn等助剂可以有效地促进Rh对CO的吸附,因此其可以促进反应活性的提高[28,33],同时维持C2含氧化合物的选择性;Li、K、V[33]等则会抑制CO的吸附,但其会使铑粒子表面烷烃生成活性位中毒。因此,虽然反应活性会降低,但其可以提高C2含氧化合物的选择性。适量Fe助剂在促进CO吸附的同时,同样可以促进非解离吸附CO的迁移,因此可以同时提高反应活性与选择性[45]。
载体的作用方式则不同,惰性载体(Al2O3、SiO2)主要通过提高铑的分散度来增加CO吸附活性位从而提高反应活性。活性载体(ZrO2、TiO2)则可以通过表面羟基物种作用调变铑粒子价态,从而改变表面Rh物种的吸附方式。不同的助剂、载体的接触方式、不同的制备方法及前体则同样可以调变铑粒子的形状价态及晶面等来改变催化剂表面CO吸附强度及种类。
6 工业应用现状及展望
铑基催化剂用于低碳醇合成反应从20世纪90年代开始就一直备受关注,其较高的醇类(特别是乙醇)选择性以及较简单的制备方法更是让人们看到了其大规模工业应用的可能性[46]。但贵金属铑昂贵的价格和较低的铑原子利用率在很大程度上限制了应用,在众多的研究学者、机构中,只有大连化学物理研究所对铑基催化剂体系进行了中试,他们制备的Mn-Li-Fe催化剂表现出了较好的活性和选择性,并具有超过1000h以上的稳定性,表现出了良好的工业应用前景。
在综述了文献中关于铑基催化剂反应机理、助剂和载体的研究后,发现不同种类的助剂、载体对铑基催化剂的活性选择性影响较大,但催化剂活性中心铑粒子是决定反应活性、选择性的基础。高活性、高选择性的活性中心铑粒子的形成是助剂、载体以及制备方法共同协同作用的结果。综合各个研究者的观点不难发现:具备适合的大小[27-28](3~6nm)、价态[21,23]、层间分布[34]及晶型的铑粒子在理论上会具有理想的反应活性及选择性。进一步的工作可以通过尝试新型助剂组合、载体以及制备方法不断修饰活性中心铑粒子,从而不断提高其反应活性与选择性,以期可以大规模地生产应用。
参考文献
[1]中国石油化工股份有限公司北京化工研究院.由合成气制低碳含氧化合物的方法:201310491393.6[P].2013 -10-18.
[2]张永青,钟炳,王琴,等. CO加氢Co催化剂研究进展[J]. 天然气化工,1997,24:49-52.
[3]FISHER F,TROPSCH H. The preparation of synthetic oil mixtures from carbon monoxide and hydrogen[J]. Brennst. Chem,1923,4:276-285.
[4]STREITWIWIESER A,Health C. Introduction to Organic Chemistry[M]. London:Collier-Macmillan,1976:279-320.
[5]HECKS R F,BRESLOW D S. The reaction of cobalt hydrotetra-carbonyl with olefins[J]. Am. Chen. Soc.,1961,83:4023-4027.
[6]ORCHIN M,RUPILIUS W. On the mechanism of the OXO reaction[J]. Catal. Rev.,1972,6(1):85-131.
[7]TAKEUCHI A,KATZER J R. Ethanol formation mechnism from CO+H2[J]. Phys. Chem.,1982,86:2438-2441.
[8]FARVE T L,VD F G,LEE V,et al. Heterogeneous catalytic insertion mechanism of the C2oxygenates formation[J]. Chem. Commun.,1985,4:230-231.
[9]ORITA H S,NAITO K,TANMARU R,et al. Mechanism of formation of C2oxygenates compounds from CO+H2reation over SiO2-supported Rh catalysts[J]. J. Catal.,1984,90:183-193.
[10]ICHIWAKA M,FUKUSHIMA T. Mechanism of syngas conversioninto C2-oxygenates such as ethanol catalysed on a SiO2-supported Rh-Ti catalysts[J]. Chem. Commun.,1985(6):321-323.
[11]OHTANI Y I,Michel E F,Gleason S. et al. Antiferro-ferromagnetic transition and micro-structural properties in a sputter deposited FeRh thin film system[J]. Appl. Phys.,1993,74:3328-3332.
[12]KAZTER J R,SLEIGHT A W,MCMEILLIAN P,et al. The role of the support in CO hydrogenation selectivity of supported rhodium[J]. Faraday Discuss,1981,72:121-133.
[13]JOHNSTON P,JOYNER R W,PUDNEY D A,et al. The interaction of synthesis gas (CO-H2) with small rhodium particles[J]. Phys.:Condens. Matter.,1989,1:SB171–SB176.
[14]MAO Wei,SU Junjie. Kinetics study of C2+oxygenates synthesis from syn-gas over Rh–MnOx/SiO2catalysts[J]. Chemical Engineering Science,2015,135:312-322.
[15]LIU J J,GUO Z,CHILDERS D,et al. Correlating the degree of metal–promoter interaction to ethanol selectivity over Mn Rh/CNTs CO hydrogenation catalysts[J]. J. Catal.,2014,313:149-158.
[16]YU J,MAO D,HAN L. Catalytic conversion of syn-gas into C2+oxygenates over Rh/SiO2-based catalysts:the remarkable effect of hydroxyls on the SiO2[J]. Mol. Catal. A:Chem.,2013,367:38–45.
[17]CHOI Y,LIU P. Mechanism of ethanol synthesis from syngas on Rh (111)[J]. Am. Chem. Soc.,2009,131:13054-13061.
[18]KAPUR N,HYUN J,SHAN B,et al. Influence of the carrier on the interaction of H2and CO with supported Rh[J]. Phys. J. Chem. C,2010,114:10171-10182.
[19]GAO Jia,MO Xunhua. Selective activation of the C—O bonds in lignocell ulosic biomass for the efficient production of chemicals[J]. Journal of Catalysis,2010,275:211-217.
[20]WASTON P R,SOMORJAI G A. The formation of oxygen-containing of organic molecules by the hydrogenation of carbon monoxide using a lanthanum Rhodate catalyst[J]. J. Catal.,1982,74(2):282-295.
[21]WASTON P R,SOMORJAI G A. The hydrogenation of carbon monoxide over Rhodium oxide surface[J]. J. Catal.,1987,103:407-418.
[22]PAN Xiulian,BAO Xinhe. Enhanced ethanol production inside carbon-nanotube reactors containing catalytic particles[J]. Natural Material,2007,35:507-511.
[23]VICTOR Abdelsayed,DUSHYANT Shekhawat,Poston J A,et al. Synthesis,characterization,and catalytic activity of Rh-based lanthanum zirconate pyrochlores for higher alcohol synthesis[J]. Catalysis Today,2013,207:65-73.
[24]VAN DEN BENG F G A,GLEZRE J H E,SACHLTER W M H. The role of promoters in CO/H2reaction:effects of MnO and MoO2in silica-supported rhodium catalysts[J]. J. Catal.,1985,93(2):340-352.
[25]KAZTER J R,SLEIGHT A W,GAJARDO P,et al. The role of the support in CO hydrogenation of supported rhodium[J]. Faraday Disc. Chem. Soc.,1981,72:121-133.
[26]陈明英,翁维正,华卫琦,等. 合成气制C2含氧化合物 Rh-Mn/SiO2催化剂上CO 吸附的红外光谱研究[J]. 催化学报,2011,32(4):672-681.
[27]HANAOKA T,ARAKAWA H. Ethylene hydroformylation and carbon monoxide hydrogenation over modified and unmodified silica supported rhodium catalysts[J]. Catalysis Today,2000,58:271-280.
[28]MANUEL Ojeda,SERGIO Rojas. Synthesis of Rh nano-particles by the microemulsion technology particle size effect on the CO + H2reaction[J]. Applied Catalysis A:General,2004,274:33-41.
[29]ICHIKAWA M. Catalysis by supportd metal crystallites from carbonyl clusters. Ⅰ.Catalytic methanol synthesis under mild conditions over supported rhodium,platinum,and iridium crystallites prepared from Rh,Pt,and Ir carbonyl cluster compounds deposited on ZnO and MgO[J]. Bull. Chem. Soc. Jpn.,1978,51:2268-2272.
[30]尹红梅. 铑基催化剂上CO加氢制备C2含氧化合物的研究[D]. 大连:中国科学院大连化学物理研究所,2003.
[31]CHUANG S S,PIEN S I. Infrared study of the CO insertion reaction on reduced,oxidized and sulfided Rh/SiO2catalysts[J]. J. Catal.,1992,135:618-634.
[32]GONZALO P,PATRICIA C,AGUSTIN M,et al. New insights into the role of the electronic properties of oxide promoters in Rh-catalyzed selective synthesis of oxygenates from synthesis gas[J]. Journal of Catalysis,2011,280:274-288.
[33]MO Xunhua,GAO Jia,UMNAJKASEAM Nattawan,et al. La,V,and Fe promotion of Rh/SiO2for CO hydrogenation:effect on adsorption and reaction[J]. Journal of Catalysis,2009,267(2):167-176.
[34]LIU Yang,LIU Ping. Ethanol synthesis from syngas on transition metal-doped Rh(111) surfaces:a density functional kinetic monte carlo study[J]. Top. Catal.,2014,57:125-134.
[35]ZHAO Yonghui,YANG Mingmei. Rh-decorated Cu alloy catalyst for improved C2oxygenate formation from syngas[J]. Phys. Chem. C,2011,115:18247-18256.
[36]TAUSTER S J,FUNG S C,GARTEN R L. Strong metal-support interaction. Group8:noble metals supported on TiO2[J]. Am. Chem. Soc.,1978,100:170-175.
[37]SMITH A K,HUGUES F,THEOLIER A. Surface-supported metal cluster on alumina,and magnesia[J]. Inong. Chem.,1979,18:3104-3112 .
[38]YU Jun,MAO Dongsen,LU Guanzhong,et al. Enhanced C2oxygenate synthesis by CO hydrogenation over Rh-based catalyst supported on a novel SiO2[J]. Catalysis Communications,2012,24:25-29.
[39]IOANNIDES T,VVERKIOS X. Influence of the carrier on the interaction of H2and CO with supported Rh[J]. Journal of Catalysis,1993,140:353-369.
[40]ICHIKAWA M. Catalysis by supported metal crystallites from carbonyl clusters[J]. Bull. Chem. Soc. Jpn.,1978,51:2273-2277.
[41]HAN Lupeng,MAO Dongsen. Synthesis of C2-oxygenates from syngas over Rh-based catalyst supported on SiO2,TiO2and SiO2-TiO2mixed oxide[J]. Catalysis Communications,2012,23:20-24.
[42]HAN Lupeng,MAO Dongsen. C2-oxygenates synthesis through CO hydrogenation on SiO2-ZrO2supported Rh-based catalyst:the effect of support[J]. Applied Catalysis A:General,2013,454:81- 87.
[43]FAN Zhongli,CHEN Wei ,PAN Xiulian. Catalytic conversion of syngas into C2oxygenates over Rh-based catalysts——effect of carbon supports[J]. Catalysis Today,2009,147:86-93.
[44]XU Boqing,SCHALTER W M H. Rh/NaY:a selective catalyst for direct synthesis of acetic acid from syngas[J]. Journal of Catalysis,1998,180:194-206.
[45]MOHAMANND A,HAIDER. Fe-promotion of supported Rh catalysts for direct conversion of syngas to ethanol[J]. Journal of Catalysis,2009,261(1):9-16.
[46]士丽敏,储伟,刘增超. 合成气制低碳醇用催化剂研究进展[J]. 化工进展,2011,30(1):162-166.
Progress on the rhodium-based catalysts for the synthesis of higher alcohol
HAN Tong,ZHAO Lin,YUE Yizhi,LIU Yuan
(School of Chemical Engineering,Tianjin University,Tianjin 300072,China)
Abstract:Literature reviews of Rh-based catalysts for higher alcohol synthesis has been made in this article. Mainly four topics of reaction mechanism,active sites,additives and carriers are discussed. We compared reports from different researchers,and found that controversies of reaction mechanism and active sites are still existed. Further researches are still needed. Though rhodium based catalysts exhibit high alcohol selectivity and good stability,its low reaction activities and high price still limit the large scale application,so efficient catalyst with high industrial applications values are still needed to study.
Key words:syngas; catalyst; alcohol; support; stability
中图分类号:TQ 174.75
文献标志码:A
文章编号:1000–6613(2016)04–1087–07
DOI:10.16085/j.issn.1000-6613.2016.04.019
收稿日期:2015-07-20;修改稿日期:2015-10-19。
猜你喜欢
分子催化(2022年1期)2022-11-02
华人时刊(2022年9期)2022-09-06
环境卫生工程(2021年2期)2021-06-09
华人时刊(2020年15期)2020-12-14
中国特种设备安全(2019年5期)2019-07-16
石油石化绿色低碳(2019年6期)2019-02-13
浙江大学学报(工学版)(2016年11期)2016-06-05
Coco薇(2016年2期)2016-03-22
中国资源综合利用(2016年4期)2016-01-22
西安交通大学学报(2014年7期)2014-04-16
