
辛基二茂铁分子结构特性的量子化学计算
2016-11-03 05:35郭志贤马晓燕王灵侠陈智群
固体火箭技术 2016年5期
郭志贤,马晓燕,王灵侠,孙 坤,高 燕,陈智群
(1.西北工业大学 理学院,陕西省高分子科学与技术重点实验室,西安 710072;2.西安近代化学研究所,西安 710065)
辛基二茂铁分子结构特性的量子化学计算
郭志贤1,马晓燕1,王灵侠1,孙坤1,高燕1,陈智群2
(1.西北工业大学 理学院,陕西省高分子科学与技术重点实验室,西安710072;2.西安近代化学研究所,西安710065)
辛基二茂铁按照取代辛烷异构可分为(1-辛基)-二茂铁、(2-辛基)-二茂铁、(3-辛基)-二茂铁和(4-辛基)-二茂铁。由于目前缺乏高纯度的各种辛基二茂铁,所以也缺乏相关物理性能数据。通过量子化学密度泛函B3LYP的方法,对4种辛基二茂铁异构体的结构进行理论计算。结果表明,气相辛基二茂铁的稳定构象中,二茂铁基团构象是重叠式,属于D5h点群,Fe可视为含有一定正电荷的离子;偶极矩分析结果表示,分子极性大小顺序为(4-辛基)-二茂铁>(3-辛基)-二茂铁>(2-辛基)-二茂铁>(1-辛基)二茂铁。用GIAO方法模拟出4种辛基二茂铁的核磁共振碳谱、氢谱,分析了辛基二茂铁中碳原子、氢原子的化学环境,为辛基二茂铁同分异构体的研究工作提供参考。
量子化学;密度泛函;B3LYP;固体推进剂;辛基二茂铁
0 引言
辛基二茂铁是典型的二茂铁烷基链一元取代衍生物,二茂铁衍生物广泛用于燃料油节油消烟剂、燃气助燃催化剂、固体火箭推进剂等领域。辛基二茂铁按照辛基取代基在茂环上的异构情况可分为正辛基二茂铁、(2-辛基)-二茂铁、(3-辛基)-二茂铁和(4-辛基)-二茂铁。工业品正辛基二茂铁纯度达90%以上,可作为丁羟复合固体推进剂的燃速催化剂[1-3],燃速催化效率高,制药工艺性能好,在航天领域广泛使用。而(2-辛基)-二茂铁、(3-辛基)-二茂铁和(4-辛基)-二茂铁来源于工业品辛基二茂铁,每种成分的含量不到25%,是通过烷基化反应一步得到的,3种物质很难分离,通常是以混合物的形式用于固体推进剂中,具体哪种成分会影响催化效率、迁移性和稳定性,目前并没有研究和报告。二茂铁基上取代基碳数不同、结构不同,均会影响其催化氧化性能,对辛基二茂铁各同分异构体物理性能进行模拟计算,对各种战略和战术武器的更高要求的研究具有重要理论意义。
DFT是用电子密度取代波函数作为研究的基本量[4-6],电子密度是可通过实验测得[7]。DFT方法已被成功应用到了分子结构和性质、电离势的计算、光谱、能谱、热化学、催化、反应机理、过渡态结构和活化势垒等问题的研究[8-9]。DFT方法的特别之处在于选择合适的交换能和相关能的形式。尽管原则上它们中的一个可选择定域密度(LSDA)形式,另一个可选择梯度形式,实际上并不协调[10-12]。由于DFT理论方面的贡献,Kohn荣获1998年诺贝尔化学奖[13]。
对二茂铁衍生物的量子化学计算,广泛采用密度泛函的方法。其中,B3LYP的方法已被验证与实验值吻合[14-16]。由于国际上尚未出现辛基二茂铁结构和性能的相关报道,通过量子化学的计算方法,对正辛基二茂铁及其同分异构体的结构进行理论研究,为日后的辛基二茂铁研究工作提供具有参考性的数据。因此,采用密度泛函B3LYP的方法,在6-31++G(d,p)基组水平上,对辛基二茂铁及其异构体构型进行优化、振动分析和极性计算;并在此基础上,用GIAO方法计算了4种辛基二茂铁的核磁共振碳谱、氢谱。
1 气相辛基二茂铁的稳定构象
二茂铁具有显著的夹心结构,具有2种特殊的构象:重叠式、交叉式。故作为其衍生物,辛基二茂铁也可能具有2种构象,从理论角度探讨辛基二茂铁4种同分异构体的稳定构象,以正辛基二茂铁为例,其可能构象如图1。
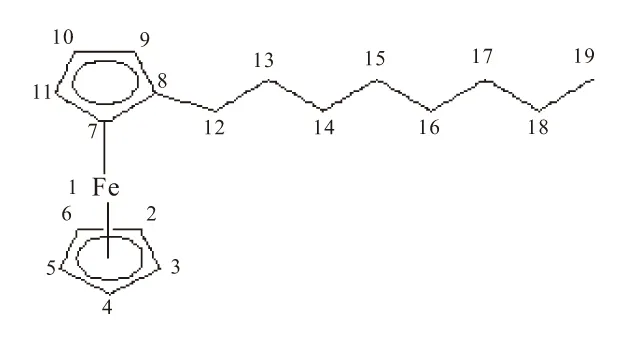
(a)重叠式
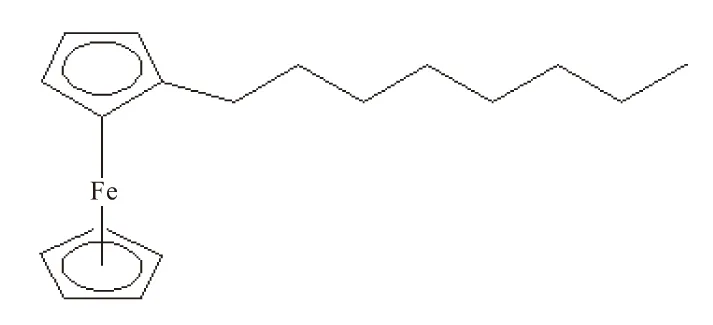
(b)交叉式
判断分子的构象是否稳定的一个重要方法是观察它的振动光谱是否出现了虚频,对正辛基二茂铁的2种构象分别用密度泛函B3LYP的方法,在6-31++G(d,p)基组水平上,对其构型进行优化,并计算和分析二者的振动频率,在计算方法和基组水平相同的前提下进行比较,结果见表1。
振动分析表明,正辛基二茂铁的重叠式构象没有虚频,而其交叉式构象出现了一个虚频,证明正辛基二茂铁的重叠式构象是稳定结构。故在气相时,正辛基二茂铁的稳定构象是重叠式构象。
重叠式正辛基二茂铁中的二茂铁基团是D5h点群对称结构,其部分键角和键长具有对应的对称性,表中数据与理论值推测相符,证明了对正辛基二茂铁的结构优化方法是合理的,说明此方法同样适用于对其异构体进行结构优化的计算。经B3LYP 6-31++G(d,p)方法优化后的4种辛基二茂铁稳定构象见图2,优化后的辛基二茂铁系列异构体的频率分析与电子空间范围见表2。
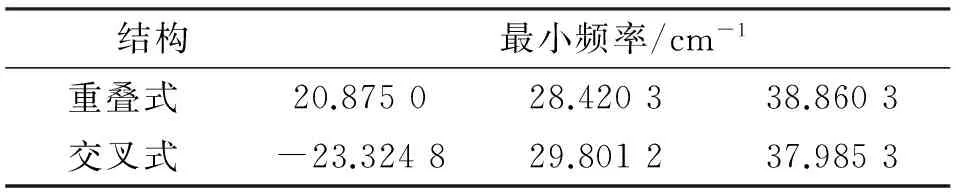
表1 正辛基二茂铁的振动分析
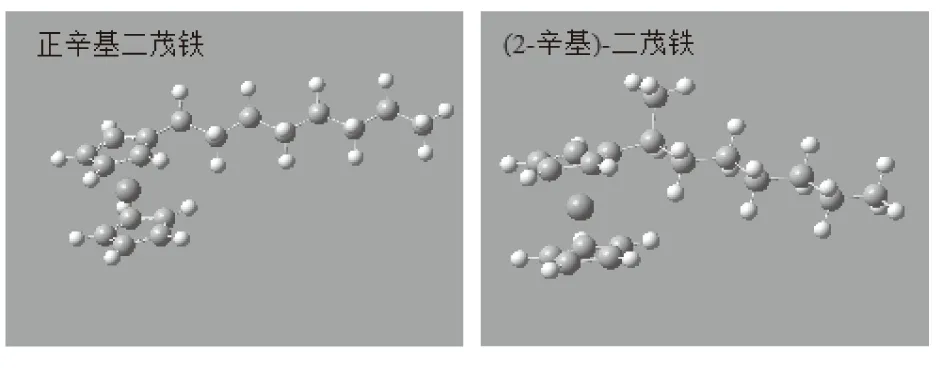
(a)正辛基二茂铁 (b)(2-辛基)-二茂铁

(c)(3-辛基)-二茂铁 (d)(4-辛基)-二茂铁
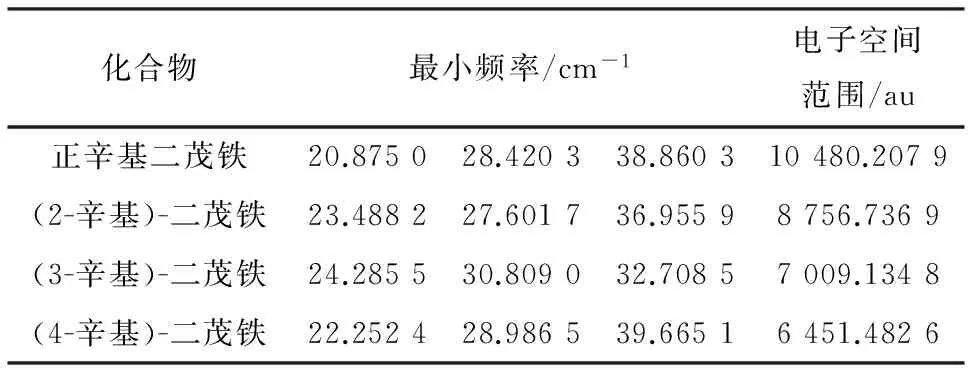
化合物最小频率/cm-1电子空间范围/au正辛基二茂铁20.875028.420338.860310480.2079(2-辛基)-二茂铁23.488227.601736.95598756.7369(3-辛基)-二茂铁24.285530.809032.70857009.1348(4-辛基)-二茂铁22.252428.986539.66516451.4826
2 辛基二茂铁的电子结构计算
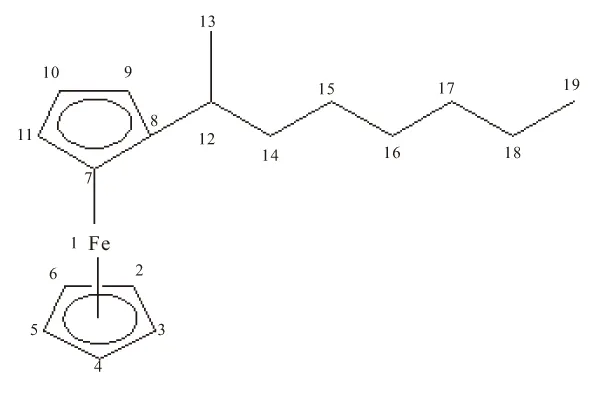
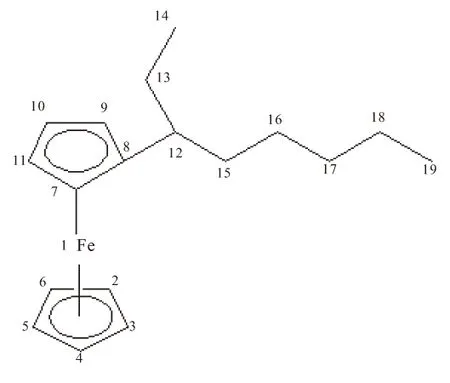
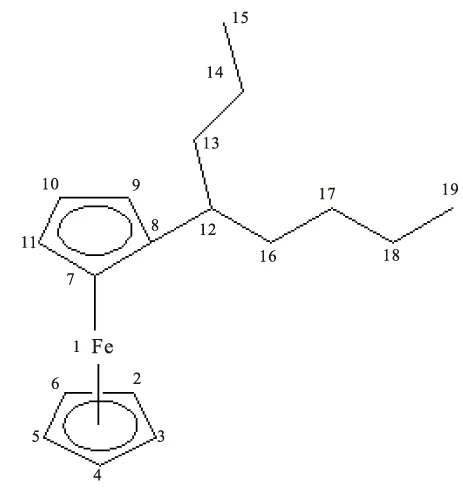
目前,文献主要认为二茂铁衍生物中的二茂铁基团是由2个环戊二烯负离子与二价铁离子形成的夹心化合物,其中的铁以二价离子态存在。通过由密度泛函计算方法,对辛基二茂铁的4种同分异构体各原子的电荷分布进行理论计算,得出了不同于以上说法的结论,假设碳元素位置如图3所示,表3列出了4种分子中各原子电荷分布的计算结果。
辛基二茂铁的合成过程中,形成的辛基二茂铁分子对电荷进行了再分配,使电荷分布发生了一系列的变化,茂环上的电荷向铁离子转移,重新分配了电荷,这种推测与分子轨道理论相符。可看出,4种异构体在8号碳原子上的带电量呈依次下降的趋势,其中正辛基二茂铁最高,说明随着辛基上支链的出现,使分子电荷发生变化,分去了茂环中8号碳原子的部分电荷;4种辛基二茂铁辛基上的—CH3含负电量最高,(2-辛基)-二茂铁的13号碳原子、(3-辛基)-二茂铁的14号碳原子、(4-辛基)-二茂铁的15号碳原子上的负电荷数明显多,表明辛基中支链端点会拥有较多电荷数,且都大于0.4的负电荷;而从8号碳原子开始到两端支链最后的—CH3之前,碳原子的带电量都是较低的;未接辛基的茂环上的电荷分布很均匀,从C2到C6,都在0.2的负电荷左右。电荷分布上的微小差异,有助于对4种同分异构体的分离工作。
(a)(2-辛基)-二茂铁
(b)(3-辛基)-二茂铁
(c)(4-辛基)-二茂铁
Table 3Charge pattern of octylferrocene isomersC/m3

原子FeC2C3C4C5C6C7正辛基二茂铁0.552-0.204-0.207-0.205-0.206-0.206-0.232(2-辛基)-二茂铁0.545-0.204-0.207-0.205-0.205-0.206-0.233(3-辛基)-二茂铁0.545-0.203-0.200-0.208-0.205-0.207-0.232(4-辛基)-二茂铁0.544-0.204-0.208-0.208-0.205-0.206-0.231原子—C8C9C10C11C12C13正辛基二茂铁—0.047-0.2370.202-0.210-0.326-0.269(2-辛基)-二茂铁—0.030-0.237-0.201-0.210-0.134-0.445(3-辛基)-二茂铁—0.020-0.233-0.202-0.200-0.125-0.251(4-辛基)-二茂铁—0.018-0.232-0.202-0.209-0.131-0.251原子—C14C15C16C17C18C19正辛基二茂铁—-0.253-0.253-0.252-0.246-0.247-0.441(2-辛基)-二茂铁—-0.275-0.260-0.253-0.246-0.247-0.441(3-辛基)-二茂铁—-0.449-0.284-0.272-0.248-0.247-0.441(4-辛基)-二茂铁—-0.254-0.442-0.284-0.266-0.249-0.442
3 辛基二茂铁的偶极矩计算
为了判断4种辛基二茂铁的分子极性大小,用密度泛函的方法,对其进行了分子偶极矩的计算,优化后的4种辛基二茂铁的偶极矩如表4所示。4种异构体的分子极性大小为(4-辛基)-二茂铁>(3-辛基)-二茂铁>(2-辛基)-二茂铁>正辛基二茂铁。可见,茂环上一元取代的辛基上支链长度影响分子的极性,支链越长,极性越大。
表44种辛基二茂铁的偶极矩
Table 4Dipole moment of octylferroceneD
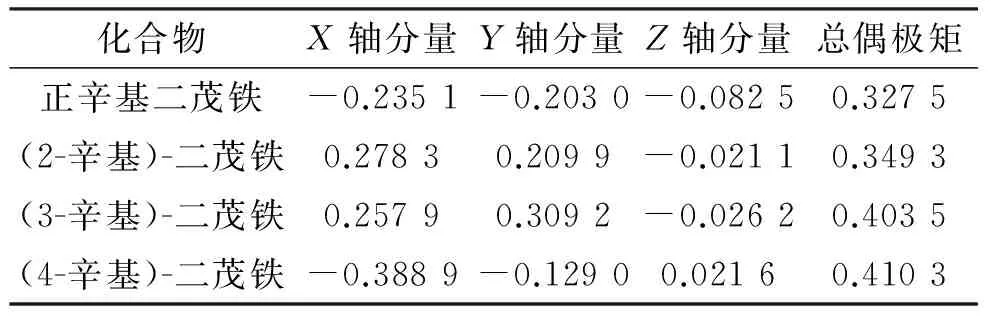
化合物X轴分量Y轴分量Z轴分量总偶极矩正辛基二茂铁-0.2351-0.2030-0.08250.3275(2-辛基)-二茂铁0.27830.2099-0.02110.3493(3-辛基)-二茂铁0.25790.3092-0.02620.4035(4-辛基)-二茂铁-0.3889-0.12900.02160.4103
4 辛基二茂铁的核磁共振碳谱、氢谱计算
经B3LYP 6-31++G(d,p)方法优化后,用密度泛函的B3LYP法,在6-31++G(d,p)基组上,用GIAO方法,计算了4种辛基二茂铁的核磁共振碳谱的化学位移。以正辛基二茂铁为例,正辛基二茂铁18个碳的化学位移计算值如表5所示,并与实验值进行了对照。结果表明,计算值与实验值的误差在8.5%以内,证明所采用的密度泛函计算方法基本合理。
由于国际上至今尚未研究出辛基二茂铁3种异构体的分离方案,使其各项理化参数空缺,经过表5的数据证实所用计算方法合理之后,对辛基二茂铁另外3种同分异构体进行了核磁共振碳谱化学位移及误差计算,设定3种辛基二茂铁碳位置如图3所示,表6给出了计算值。

表5 正辛基二茂铁核磁共振碳谱的化学位移及误差

表6 辛基二茂铁3种异构体的化学位移
从4种辛基二茂铁的碳谱可看出,3种同分异构体8号碳原子的化学位移均比正辛基二茂铁高。可见,辛基上出现支链改变了茂环上与辛基相连的碳原子的化学环境,而茂环上剩余的9个碳原子化学环境未发生变化,化学位移不变;(2-辛基)-二茂铁的13号碳原子、(3-辛基)-二茂铁的14号碳原子、(4-辛基)-二茂铁的15号碳原子均为相应分子上辛基支链的端点,其化学位移明显降低,表明辛基链发生异构,其支链端点的化学环境会发生显著变化。
对辛基二茂铁4种同分异构体进行了核磁共振氢谱计算,以下分别是计算出的4种辛基二茂铁的核磁图谱。从模拟图可看出δ4.0附近的区域表示了二茂铁基上氢原子的化学位移,而其他位置氢原子化学位移有所不同。
正辛基二茂铁的氢谱如图4所示,δ2.5附近的c区域为最靠近二茂铁基上的2个碳原子上的4个氢原子的化学位移,δ1.4附近e区域为辛基上甲基氢原子的化学位移。δ2.0附近表示了剩余—CH2—的氢原子化学位移。
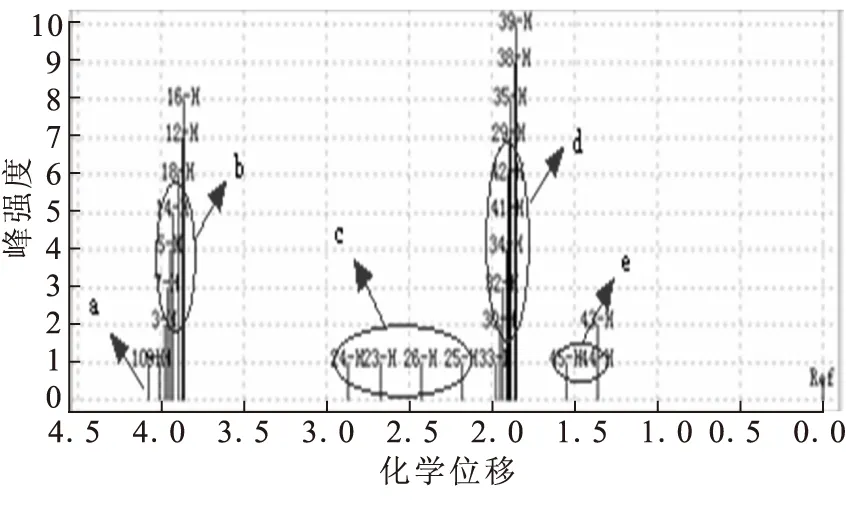
图4 正辛基二茂铁的氢谱
(2-辛基)-二茂铁的氢谱如图5所示,δ2.5附近d区域为最靠近二茂铁基上的2个碳原子上的3个氢原子的化学位移,δ1.4附近f区域为2个甲基氢原子的化学位移。δ2.0附近表示了剩余—CH2—的氢原子化学位移。
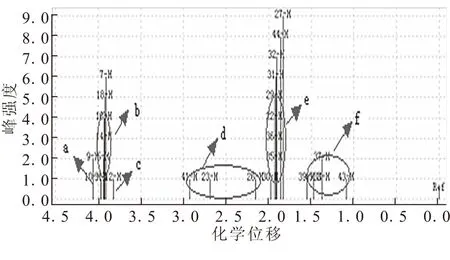
图5 (2-辛基)-二茂铁的氢谱
(3-辛基)-二茂铁氢谱如图6所示,δ2.5附近d区域为最靠近二茂铁基上的3个碳原子上的氢原子的化学位移,δ1.5附近f和g区域为2个甲基氢原子的化学位移。δ2.0附近表示了剩余—CH2—的氢原子化学位移。
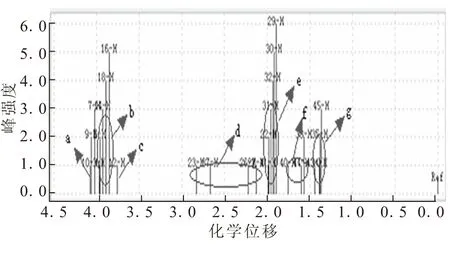
图6 (3-辛基)-二茂铁的氢谱
(4-辛基)-二茂铁的氢谱如图7所示,δ2.7附近d区域为辛基上亚甲基氢原子的化学位移,δ1.5附近g和h区域为2个甲基氢原子的化学位移。δ2.0附近表示了剩余—CH2—的氢原子化学位移。
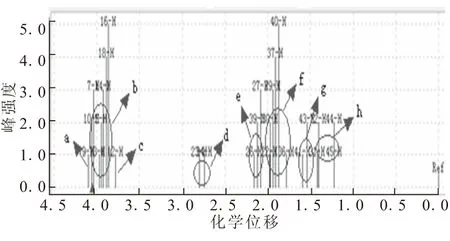
图7 (4-辛基)-二茂铁的氢谱
5 结论
采用量子化学中的密度泛函法,对辛基二茂铁系列进行了结构优化、频率计算和核磁共振碳谱计算;通过理论上的分析及部分参数与实验值的对照,证明了密度泛函B3LYP 6-31++G(d,p)方法对辛基二茂铁分子计算的可行性。量子化学计算结果表明:
(1)气相时辛基二茂铁的稳定构象中二茂铁基团是重叠式,属于D5h点群,Fe可视为含有一定正电荷的Feδ+离子。
(2)计算了4种辛基二茂铁的偶极矩,结果显示,分子极性大小为(4-辛基)-二茂铁>(3-辛基)-二茂铁>(2-辛基)-二茂铁>正辛基二茂铁,可见茂环上一元取代的辛基上支链长度影响分子的极性,支链越长,极性越大。
(3)对4种辛基二茂铁的核磁共振碳谱、氢谱进行了计算,分析了辛基二茂铁碳原子、氢原子化学环境的变化,为辛基二茂铁同分异构体的在固体火箭推进技术上的研究工作提供了具有参考性的数据。
[1]郭建勋.二茂铁-优良的燃料燃速催化剂[J].陕西化工,1995(1):35-38.
[2]张国涛,周遵宁,张同来.固体推进剂含能催化剂研究进展[J].固体火箭技术,2011,34(3):319-323.
[3]黄根龙,唐松青,丁宏勋.国外二茂铁燃速催化剂研究的新进展[J].固体火箭技术,1989(2):72-76.
[4]Hohenberg P,Kohn W.Inhomogeneous electron gas[J].Physical Review,1964,136(3B):B864.
[5]Thrower P,Sorg D 24.The pyrolysis of propylene over graphite substrates[J].Carbon,1973,11(6):672.
[6]Kohn W,Sham L J.Self-consistent equations including exchange and correlation effects[J].Physical Review,1965,140(4A):A1133.
[7]Becke A D.Density-functional exchange-energy approximation with correct asymptotic behavior[J].Physical Review A,1988,38(6):3098.
[8]Becke A D.Density-functional thermochemistry.III.The role of exact exchange[J].The Journal of Chemical Physics,1993,98(7):5648-5652.
[9]Gill P M.A new gradient-corrected exchange functional[J].Molecular Physics,1996,89(2):433-445.
[10]Grimme S.Density functional theory with London dispersion corrections[J].Wiley Interdisciplinary Reviews:Computational Molecular Science,2011,1(2):211-228.
[11]Cohen A J,Mori-Sánchez P,Yang W.Challenges for density functional theory[J].Chemical Reviews,2011,112(1):289-320.
[12]NØrskov J K,Abild-Pedersen F,Studt F,et al.Density functional theory in surface chemistry and catalysis[J].Proceedings of the National Academy of Sciences,2011,108(3):937-943.
[13]Adamo C,Barone V.Implementation and validation of the Lacks-Gordon exchange functional in conventional density functional and adiabatic connection methods[J].Journal of Computational Chemistry,1998,19(4):418-429.
[14]Mohammadi N,Ganesan A,Chantler C T,et al.Differentiation of ferrocene D 5d and D 5h conformers using IR spectroscopy[J].Journal of Organometallic Chemistry,2012,713:51-59.
[15]Lourderaj U,Venkatasubbaiah K,Sharma N,et al.Mechanisms and dynamics of protonation and lithiation of ferrocene[J].Physical Chemistry Chemical Physics,2015,17(34):22204-22209.
[16]Islam S,Wang F.The delectrons of Fe in ferrocene:the excess orbital energy spectrum (EOES)[J].RSC Advances,2015,5(16):11933-11941.
(编辑:刘红利)
Quantum chemistry calculations of octylferrocene isomers
GUO Zhi-xian1,MA Xiao-yan1,WANG Ling-xia1,SUN Kun1,GAO Yan1,CHEN Zhi-qun2
(1.The Key Laboratory of Polymer Science and Technology,School of Science,Northwestern Polytechnical University, Xi'an710072,China;2.Xi'an Modern Chemistry Research Institute,Xi'an710065,China)
Octylferrocene isomers could be divided into n-octylferrocene,(2-octyl)-ferrocene,(3-octyl)-ferrocene and (4-octyl)-ferrocene.High purity of n-octylferrocene,(2-octyl)-ferrocene,(3-octyl)-ferrocene and (4-octyl)-ferrocene had never been obtained categorically.In this study,quantum chemistry calculations on octylferrocene were researched.DFT/B3LYP calculations were carried out for structure optimization,vibration analysis and polarity calculations of octylferrocene.The results show that stable conformation of ferrocene group is overlapped and belongs to the D5h group in gas phase.‘Fe’ could be seen as a positively charged ion;the sequence of molecular polarity is (4-octyl)-ferrocene>(3-octyl)-ferrocene>(2-octyl)-ferrocene>n-octylferrocene.On the basis,GIAO was used to calculate 13C-NMR and 1H-NMR of octylferrocene.
quantum chemistry;density functional;B3LYP;solid propellant;octylferrocene
2015-07-27;
2015-08-25。
国防科工局火炸药技术基础研究项目(20141481)。
郭志贤(1989—),硕士生,研究方向为辛基二茂铁分离纯化研究。E-mail:zhixian1888@163.com
马晓燕(1963—),女,博士生导师,主要从事辛基二茂铁分离纯化研究。E-mail:m_xiao_yana@nwpu.edu.cn
V512
A
1006-2793(2016)05-0672-06
10.7673/j.issn.1006-2793.2016.05.014
猜你喜欢
物理学报(2022年10期)2022-06-04
中学生数理化(高中版.高考理化)(2022年2期)2022-04-26
中学生数理化(高中版.高二数学)(2020年6期)2020-07-24
中小学班主任(2019年12期)2019-09-10
考试周刊(2018年15期)2018-01-21
中学化学(2017年5期)2017-07-07
新课程·中旬(2016年12期)2017-05-08
教育教学论坛(2017年14期)2017-04-20
中学生数理化·高二版(2017年2期)2017-04-19
湖北农业科学(2012年16期)2012-09-12
