
类石墨烯C3N4纳米片光催化分解水制氢中的量子限域效应
2016-11-22 09:49郝旭强靳治良敏世雄吕功煊
物理化学学报 2016年10期
郝旭强 杨 浩 靳治良,* 续 京 敏世雄 吕功煊
(1北方民族大学化学与化学工程学院,银川750021;2中国科学院兰州化学物理研究所,羰基合成与选择氧化国家重点实验室,兰州730000)
类石墨烯C3N4纳米片光催化分解水制氢中的量子限域效应
郝旭强1杨浩1靳治良1,*续京1敏世雄1吕功煊2,*
(1北方民族大学化学与化学工程学院,银川750021;2中国科学院兰州化学物理研究所,羰基合成与选择氧化国家重点实验室,兰州730000)
从层状化合物获得的纳米片是一类新型纳米结构材料,这种二维各向异性的纳米甚至亚纳米级的材料具有独特的物理化学性能,其中最好的一个例证就是从石墨烯C3N4到石墨烯C3N4纳米片的转变。通过高温氧化热刻蚀方法将体相g-C3N4剥离成g-C3N4纳米片,应用于染料敏化可见光分解水产氢,表现出了较体相g-C3N4高于2.6倍的产氢速率。通过X射线衍射(XRD)、傅里叶变换红外(FTIR)光谱、扫描电子显微镜(SEM)、Brunauer-Emmett-Teller(BET)、荧光光谱和光电化学等表征研究了g-C3N4纳米片的结构及曙红(EY)和g-C3N4纳米片之间的电子迁移过程。热剥离后的g-C3N4纳米片具有较高的比表面积,不仅可以更为有效地吸附染料分子,还因其量子限域效应大大增强了光生电荷的分离效率和电子转移效率,改善了电子沿平面方向的传输能力以及光生载流子的寿命,从而显著提高g-C3N4纳米片的光催化产氢活性。
g-C3N4纳米片;染料敏化;量子限域效应;光催化;产氢
1 引言
近年来,光催化制氢成为太阳能转化与储存的热点1-3,其中新型的有机聚合物半导体光催化剂——石墨烯氮化碳(g-C3N4),由于具有可见光响应、较高的稳定性等优点,使其成为光催化技术领域的研究热点4-7。
石墨烯氮化碳是一种直接带隙半导体,理想条件下室温禁带宽度为2.7 eV左右,其具有与碳材料相似的层状堆积结构和sp2杂化的π共轭电子能带结构8,9。虽然g-C3N4具有优良的化学稳定性、热稳定性和可见光催化活性,但由于其比表面积较小,带隙宽度相对较大,对可见光响应的范围较小,光生电荷分离程度不高且容易复合等缺陷,严重制约了其在光催化领域和能源领域的广泛应用10-12。研究表明,通过提高g-C3N4的比表面积、掺杂以及与其他半导体复合等手段能够提高其对可见光的响应范围和提高其光生电子与空穴的分离效率13-18。
石墨烯C3N4纳米片相比于体相g-C3N4具有更高的比表面积和较大的禁带宽度,是一种新型的纳米级材料。其具有低的纳米尺寸甚至亚纳米尺寸厚度以及二维各向异性等特点,可以提高电子转移效率,并且由于其具有量子限域效应(quantum confinement effect)能够增强光激发载流子寿命相比于体相g-C3N4,因此能够显著提高其光催化效率19,20。例如:Cheng等21通过超声辅助液态剥离方法将大体积g-C3N4制备为g-C3N4纳米片,然后通过光还原将Au纳米粒子(AuNPs)负载到g-C3N4纳米片上,在可见光下降解甲基橙表现出优异的光催化性能。Cao等22通过高温加热制备g-C3N4纳米片光催化剂,通过水热法将CdS量子点原位生长到g-C3N4纳米片表面表现出很高的可见光催化产氢活性,g-C3N4纳米片/12%(w,质量分数)CdS光催化剂的产氢速率为4.494 mmol·h-1·g-1,是g-C3N4的115倍高。研究表明,光催化产氢活性的增强是由于CdS和g-C3N4纳米片界面间光生电子转移效率提高。Tonda等23通过浸渍沉积制备Fe掺杂g-C3N4纳米片光催化剂,2%(x,摩尔分数)Fe掺杂g-C3N4纳米片光催化剂对降解罗丹明B表现出很高的可见光催化活性,是g-C3N4光催化活性的7倍高,并且经过5次循环后仍具有很高的光催化稳定性。研究表明,Fe掺杂g-C3N4纳米片具有高的可见光响应能力、大的比表面积、高的电荷分离效率和电子转移效率,因而有效地增强了光催化活性。Lotsch等24也发现将g-C3N4制备晶体g-C3N4纳米片显著地增强了其可见光催化产氢活性。
染料敏化是一种很有效的拓宽半导体可见光吸收范围和吸收强度的方法,并且在染料敏化太阳能电池(DSSC)方面获得了很大的成功25,26。因此,染料敏化在光催化分解水制氢方面也被广泛地研究27-36。Takanabe等31首先研究了染料敏化g-C3N4的光催化产氢活性,他们将酞菁镁染料沉积于介孔C3N4(MgPc/mpg-C3N4)表面有效地拓展了光吸收范围,表现出稳定的光催化产氢活性,在660 nm时其表观量子效率为0.07%。Xu等33在不同温度(450-650°C)下煅烧尿素制备了不同的g-C3N4,并研究了其曙红(EY)敏化的产氢活性,EY敏化g-C3N4(600°C)的表观量子效率达到18.8%。而对于染料敏化g-C3N4纳米片的报道很少,Wang等34以赤藓红B(ErB)为敏化剂研究了g-C3N4纳米片的光催化剂产氢活性,其产氢速率可达652.5 mmol· h-1,460 nm波长下其量子效率可达33.4%。因此,染料敏化是提高催化剂可见光催化活性的有效手段。
本文通过高温热氧化将体相g-C3N4剥离成g-C3N4纳米片材料,进一步提高了其比表面积、带隙宽度和电子传导性能。以曙红为光敏化剂、三乙醇胺(TEOA)为电子给体、Pt为产氢助催化剂,研究了染料敏化g-C3N4纳米片的光催化产氢活性。通过X射线衍射(XRD)、傅里叶变换红外(FTIR)光谱对催化剂的结构进行表征,并通过对光催化体系产氢荧光光谱以及光电化学性能测试,分析了EY和g-C3N4纳米片之间的电子迁移过程,在此基础上提出了染料敏化g-C3N4纳米片催化剂高效光催化分解水产氢的反应机理。
2 实验部分
2.1试剂与仪器
曙红(分析纯,天津市光复科技发展有限公司)、三聚氰胺(分析纯,天津市光复科技发展有限公司);浓HCl(分析纯,36%-38%,天津市光复科技发展有限公司);氯铂酸(分析纯,沈阳市科达试剂厂);三乙醇胺(分析纯,天津市光复科技发展有限公司),所有试剂使用前未经进一步纯化处理。XRD晶相分析采用D/max RB型X射线粉末衍射仪(日本电子公司;Cu Kα,管电流30 mA,管电压40 kV);红外光谱采用Spectrum100傅里叶变换红外光谱仪(美国PerkinElmer公司)测定;催化剂的高分辨率透射电子显微镜(HRTEM)表征采用Tecnai F-30FEG型高分辨透射电镜(FEI公司,美国),加速电压为300 kV;高分辨扫描电镜(HRSEM)照片利用日本电子株式会社的JEOL JSM-6701F型扫描电子显微镜对样品进行微观形貌观察,加速电压为5 kV,样品滴加在铜托上,为了提高样品的导电性能,进行喷金处理;光电子能谱(XPS)采用英国VG Scientific ESCALAB 250Xi型光电子能谱仪测定(Mg靶,C 1s校正到285.00 eV);紫外-可见漫反射光谱(带隙)采用UV-2550 (shimadzu)光谱仪测定(BaSO4作为参比);催化剂的比表面积、孔径和孔容是在Micromeritics ASAP 2020吸附仪上进行测试;荧光光谱采用Fluoro MAX-4光谱仪(法国HORIBA Scientific公司)分析荧光强度。
2.2石墨烯氮化碳纳米片的制备
石墨烯氮化碳纳米片(g-C3N4-NS)制备方法:以三聚氰胺为原料,在空气气氛下一步热解得到了g-C3N4,然后继续加热氧化,其具体合成步骤如下:取10 g三聚氰胺放入带盖的坩埚中,在马弗炉中于550°C煅烧4 h,冷却到室温后,得到亮黄色的类石墨烯氮化碳g-C3N4,研磨成粉末后备用。然后称取400 mg g-C3N4粉末放入带盖的坩埚中,在马弗炉中于550°C继续煅烧2 h,得到g-C3N4纳米片(g-C3N4nanosheet,简写为g-C3N4-NS)光催化剂18。
2.3光催化分解水产氢实验和量子效率测定
光催化分解水产氢实验在带平面窗口的石英反应瓶(162 mL)中进行,光源为300 W氙灯,配有420 nm截至滤光片。具体步骤如下:将30 mg g-C3N4粉末加入到100 mL 10%(φ,体积分数)TEOA溶液中,超声分散10 min后向体系中依次加入一定量的1 mg·mL H2PtCl6溶液和曙红,用磁力搅拌器搅拌。开始光照前,体系通氮气置换40 min以排除瓶中空气,光照过程中用磁力搅拌器持续搅拌,使催化剂保持悬浮状态,每隔30 min从反应瓶中抽取0.5 mL的气体,用气相色谱仪(GC7900,上海天美仪器公司)分析产生的H2量(热导检测器,高纯氮气为载气,13X分子筛填充柱),外标法定量。
在测定产氢表观量子效率(AQE)时,氙灯配有不同波长的通带滤光片(430、460、490、520和550 nm)。采用经过标定的FU100型辐射计(硅光检测器,波长范围400-700 nm,灵敏度为10-50 μV·μmol-1·m-2·s-1)测定入射光子数,平行测定多个数据点,取平均值。测量入射光照射到反应器平面窗口的有效面积,通过公式(1)计算出入射光子数(np),其中,t为时间(单位为s或h),S为反应器的有效光照面积(m2),Q为辐射计测定的入射光子通量数(μmol-1·m-2·s-1)

按照公式(2)计算产氢AQE时,前提是假定入射的光子全部被体系吸收,并且不做任何的折射散射校正,其中n(H2)为氢气的产量(μmol)。

2.4工作电极的制备和光电性能测试
工作电极采用物理沉积法制备:取一定量的催化剂粉末超声分散在乙醇中形成泥浆状。取一定体积的泥浆状催化剂,滴加于氧化铟锡(ITO)透明导电膜玻璃表面,再将500 μL的曙红溶液滴到电极的催化剂薄膜表面,干燥后得到负载有催化剂的ITO工作电极。光电化学实验在美国普林斯顿电化学工作站(PAR Versa STAT 4)上进行,采用标准的三电极体系进行分析。对电极为Pt片电极,参比电极为饱和甘汞电极(SCE),电解液为0.1 mol·L-1Na2SO4和10%(φ)TEOA混合水溶液,工作电极在电解液中的浸泡面积为0.95 cm2,光源为300 W氙灯,用420 nm的截止滤光片滤去紫外光。
3 结果与讨论
3.1石墨烯氮化碳纳米片的结构表征分析
图1(A)为高温热氧化体相g-C3N4前后的XRD表征结果。从图中可以看出,体相g-C3N4具有六方相g-C3N4(JCPDS#87-1526)的特征,有两个特征衍射峰。其中,2θ=27.48°的强衍射峰对应于g-C3N4的(002)晶面,其层间距d=0.336 nm,归属于π共轭平面的类石墨烯层状堆积;另一衍射峰2θ=13.1°对应于g-C3N4的(100)晶面,是七嗪环(C6N7)重复结构基元构成的,晶面间距为d=0.765 nm,这与文献1报道结果一致。当体相g-C3N4热氧化制备成g-C3N4纳米片后,其仍具有2个特征衍射峰,分别为13.1°和27.56°。从图中可以看出热氧化后27.48°位置的衍射峰偏移到了27.56°,向右偏移了0.08°,表明π共轭堆积的平面类石墨烯层间的晶面间距减小,这与文献19报道结果一致。

图1 (a)g-C3N4和(b)g-C3N4-NS的(A)XRD衍射图和(B)傅里叶变换红外光谱图Fig.1 (A)XRD patterns and(B)FTIR spectra of(a)bulk g-C3N4and(b)g-C3N4-NS
红外表征结果进一步证实了g-C3N4-NS的晶体结构,如图1(B)所示,从图中可以看出g-C3N4和g-C3N4-NS的红外波谱图基本相似。在808 cm-1处的尖峰为g-C3N4结构中的七嗪(C6N7),从900到1800 cm-1位置的峰属于三角形的C―N(―C)―C和C―NH―C桥键结构单元,这些峰变得尖锐是由于高温热氧化层蚀刻后使得氢键结合的长链聚合物单元变得更有序,而3000到3600 cm-1之间的宽峰属于N―H伸缩键。
图2(A,B)为C3N4热氧化剥离前后的扫描电镜(SEM)照片,从图2(A)中可以清晰地看出体相g-C3N4为片层状结构,这些片状结构紧密堆叠在一起形成块体,在其表面可以观察到有许多微孔。从图2(B)中可以观察到经热氧化剥离后的g-C3N4-NS为小片层状结构,这些小片层近似颗粒状,与体相g-C3N4相比较为分散。图2(C,D)为体相g-C3N4的透射电镜(TEM)照片,图2(E,F)为热氧化剥离后的g-C3N4-NS的TEM照片。从照片可以看出体相g-C3N4为相互堆叠的片层状结构,g-C3N4-NS为小片层状结构,说明高温热氧化途径能有效地改性体相g-C3N4的结构。

图2 (A)体相g-C3N4和(B)g-C3N4-NS的扫描电镜(SEM)照片;(C,D)体相g-C3N4和(E,F)g-C3N4-NS的透射电镜(TEM)照片Fig.2 SEM images of(A)bulk g-C3N4and(B)g-C3N4-NS;TEM images of(C,D)bulk g-C3N4and(E,F)g-C3N4-NS
图3为g-C3N4-NS催化剂的XPS图谱,进一步确定了其化学组成和元素化学态。图3(A)中C 1s具有两个单峰,其结合能分别为284.7和288.4 eV。前一个峰为标准的碳峰,而后一个峰归属于g-C3N4晶格中与3个N原子相连的碳原子。图3(B)为N 1s的精细谱,曲线拟合后出现3个峰,其结合能分别为398.8、400.28和401.5 eV,分别代表着g-C3N4中C=N―C、N―(C)3和C―N―H官能团中的N原子37。

图3 g-C3N4纳米片的(A)C 1s和(B)N 1s的XPS精细谱Fig.3 (A)C 1s and(B)N 1s XPS scan spectra of g-C3N4-NS
图4为热剥离前后C3N4的UV-Vis漫反射光谱(带隙),从图中可以看出体相g-C3N4和g-C3N4-NS带隙分别为2.68和2.77 eV,表明热剥离后进一步拓宽了体相g-C3N4的带隙,减小了电子-空穴复合几率,延长了光生载流子的寿命。

图4 体相g-C3N4和g-C3N4-NS的UV-Vis漫反射光谱Fig.4 UV-Vis diffuse reflectance spectra of bulk g-C3N4and g-C3N4-NS
图5(A)为体相g-C3N4和g-C3N4-NS样品的氮气吸附-脱附等温线(BET),从图中可以看出两组样品呈现III型结构H3型回滞环,其孔径分布如图5(B)所示,体相g-C3N4和g-C3N4-NS样品的孔尺寸分别为34.96和21.87 nm,该催化剂为介孔类材料。从表1中g-C3N4和g-C3N4-NS样品的比表面积、孔体积、孔径等参数可以看出,热剥离后生成的g-C3N4-NS相比于体相g-C3N4具有更大的比表面积和孔体积,该特性更有利于染料分子的吸附,从而增强了g-C3N4-NS的催化活性。

图5 (A)体相g-C3N4和g-C3N4-NS的N2吸脱附等温线及(B)孔径分布图Fig.5 (A)Isotherms and(B)pore size distributions of bulk g-C3N4and g-C3N4-NS
3.2EY敏化g-C3N4-NS/Pt在可见光下的光催化产氢活性和稳定性

表1 催化剂的N2等温吸附表征Table 1 N2adsorption isotherms characterization of catalysts
图6(A)为体相g-C3N4和g-C3N4-NS催化剂的可见光催化产氢活性。从图中可以看出EY-g-C3N4-NS/1%(w)Pt的光催化活性明显高于体相g-C3N4/ 1%(w)Pt催化剂。在可见光下,曙红敏化g-C3N4/ Pt催化剂5 h的产氢量仅为439.79µmol,表明体相g-C3N4的光催化产氢活性低,其原因是由于其本身低的光生电子-空穴对分离效率,这使得其不利于质子在其表面还原。而EY敏化g-C3N4-NS/Pt催化剂在相同时间内的产氢量是1143.48µmol,相比于体相g-C3N4/Pt催化剂产氢活性提高了2.6倍。这是因为体相g-C3N4中非金属元素易氧化,在高温热氧化过程中,氮原子首先被氧化形成氮空位,其次是碳原子,且这种氧化始于块体表面逐步向体相推进,这使得g-C3N4-NS相比于体相g-C3N4具有更高的比表面积、更大的带隙、更长的光生载流子寿命和更好的电子传导性能19。这些特性使得g-C3N4纳米片具有优异的光催化性能,在可见光下光解水产氢速率相比于体相g-C3N4明显提高。

图6 (A)EY敏化g-C3N4/1%(w)Pt和g-C3N4-NS/1%(w)Pt的可见光产氢活性;(B)EY-g-C3N4-NS/1%(w)Pt催化剂的产氢稳定性测试Fig.6 (A)Photocatalytic activities of EY-sensitized g-C3N4/ 1%(w)Pt and g-C3N4-NS/1%(w)Pt for hydrogen evolution under visible light irradiation;(B)stability testing of H2evolution over EY-g-C3N4-NS/1%(w)Pt photocatalyst
图6(B)为EY-g-C3N4-NS/1%Pt催化剂的产氢稳定性测试。从图中可以看EY-g-C3N4-NS/1%Pt表现出良好的光催化产氢稳定性。第1个产氢循环产氢量为1143.48µmol,第二个产氢循环为1008 µmol,第三个产氢循环为891.1µmol,第四个产氢循环为629.7µmol。第五个循环重新加入曙红后产氢量为887.1µmol,第六个循环产氢量为680.7 µmol。从图中可以看出产氢量随着光照时间的增加逐渐下降,其主要原因可能是随着反应时间的增加EY发生了降解,导致产氢活性下降;经重新加入染料后,其产氢活性有一定恢复,但并未能达到初始状态,说明在该反应体系中,染料的降解是导致产氢活性降低的主要因素,但并不是唯一原因。
3.2.1g-C3N4-NS/Pt光催化剂的结构表征
为了探讨产氢稳定性测试过程中光催化性能下降的原因,对反应之后的g-C3N4-NS/Pt进行了SEM、TEM和XRD表征。图7(A)为催化剂的SEM照片,很明显,经过较长时间的产氢循环反应后g-C3N4-NS发生了较严重的团聚,这也可能是光催化性能下降的原因之一。从TEM图(图7(B))中可以看出经过较长时间的循环反应后虽然在g-C3N4-NS表面生成了Pt纳米粒子,但其分布并不均匀,这也可能是光催化性能下降的原因之一;图7(C)中可以清晰看到Pt的晶格条纹,其晶格间距为d= 0.226 nm,归属于Pt(111)晶面。图7(D)为催化反应前后的XRD图谱,从图中可以看出g-C3N4-NS/Pt在2θ=27.56°的衍射峰强度相比于反应前发生下降,说明反应后g-C3N4-NS的结晶度降低,这可能也是光催化性能下降的原因。此外,在XRD图谱中未观察到Pt的存在,这是由于加入的氯铂酸量很少的原因。
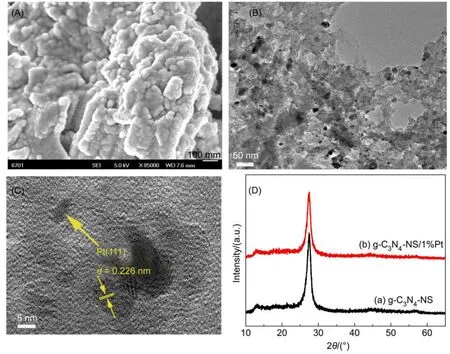
图7 反应后g-C3N4-NS/Pt光催化剂的(A)扫描电镜(SEM)、(B,C)透射电镜(TEM)照片及(D)g-C3N4-NS和g-C3N4-NS/1%Pt的XRD图谱Fig.7 (A)SEM,(B,C)TEM images of g-C3N4-NS/Pt photocatalyst after reaction and (D)XRD patterns of g-C3N4-NS and g-C3N4-NS/1%Pt
3.2.2光催化析氢过程中活性物种EY的表征
图8为体相g-C3N4/1%Pt和g-C3N4-NS/1%Pt存在时曙红浓度在可见光下的变化及降解EY的一级反应动力学曲线(实验条件与光催化产氢条件一致)。从图8(A)的暗反应中可以看出g-C3N4-NS/1% Pt对染料的吸附性能强于体相g-C3N4/1%Pt催化剂,这是由于g-C3N4-NS具有更大的比表面积和孔体积,有利于染料的吸附;当光照时,曙红浓度随着光照时间的增加不断降解,这说明催化反应过程中染料在不断的降解,这验证了稳定性测试过程中活性物种EY的降解是光催化活性下降的主要原因的推测。光降解反应遵循一级反应动力学方程:-ln(C/C0)=kt(C和C0分别代表任意时刻t和初始时刻EY的浓度),从图8(B)中可以看出g-

图8 (A)体相g-C3N4/1%Pt和g-C3N4-NS/1%Pt存在时曙红浓度在可见光下的变化及(B)降解EY的一级反应动力学曲线Fig.8 (A)Concentration changes of EY under visible light in the presence of bulk g-C3N4/1%Pt and g-C3N4-NS/1%Pt and (B)the first-order reaction kinetics curves of EY degradation
C3N4
1%P C3N4的下3.3 -NS/1%Pt降解EY的速率明显大于体相g-C3N4/ t,进一步说明了g-C3N4-NS的活性优于体相g-。综上原因,导致了循环产氢后期催化剂活性降。
EY敏化g-C3N4-NS/Pt光催化产氢条件的优化
3.3.1pH和EY浓度对g-C3N4-NS/Pt光催化产氢活性的影响
图9为EY-g-C3N4-NS/1%Pt体系在不同pH条件下的产氢活性。从图中可以看出pH值显著影响染料敏化光催化产氢体系的效率,酸性和强碱条件不利于产氢反应进行。从图9可以看出,体系pH从5到8,产氢速率逐渐增加,在pH=8时产氢速率最快,达到282.92 μmol·h-1。这可能是由于pH值影响TEOA的存在形态,并且在pH 8时溶液中TEOA可以完全还原猝灭3(EY2-)*,同时染料可以有效地吸附在催化剂表面。此外,pH从8到10可以看出产氢速率急剧下降,从282.92 μmol·h-1降到21.56 μmol·h-1,这是因为高pH值导致溶液中低的质子浓度,同时不利于H+质子的还原。
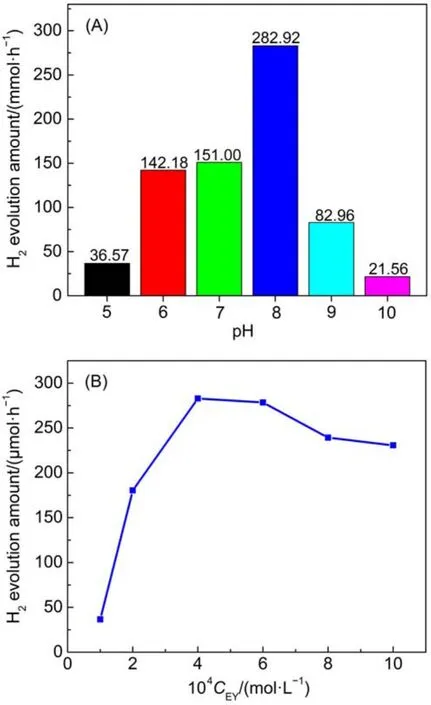
图9 (A)pH和(B)EY浓度对EY-g-C3N4-NS/1%Pt光催化产氢活性的影响Fig.9 Effect of the(A)pH and(B)EY concentration on the photocatalytic H2evolution over EY-g-C3N4-NS/1%Pt
图9(B)为EY浓度变化对EY-g-C3N4-NS/1%Pt体系光催化产氢活性的影响。从图中可以看出,在EY浓度低于4.0×10-4mol·L-1时,EY敏化g-C3N4-NS/1%Pt体系产氢速率随着EY浓度增加而增加,从36.56 μmol·h-1(CEY=1.0×10-4mol·L-1)增加到282.92 μmol·h-1(CEY=4.0×10-4mol·L-1);而EY浓度高于4.0×10-4mol·L-1时,体系的光催化产氢速率随EY浓度的升高而略微下降,从282.92 μmol·h-1(CEY=4.0×10-4mol·L-1)降到230.61 μmol·h-1(CEY=1.0×10-3mol·L-1)。这可能是由于游离染料的屏蔽效应(shielding effect),在较低EY浓度下,随着EY浓度的增加,催化剂表面染料吸附浓度也增加,这有利于提高催化剂表面染料对可见光的吸收效率,以及光生电子向半导体表面的注入,因此在较低浓度下产氢活性会随着曙红浓度的增加而升高;然而,当EY浓度过高时,不仅减少了照射到催化剂表面染料的光量子数,而且体系中游离态的EY因吸收大部分入射光而被激发且极易发生猝灭,降低了光电子的迁移效率,从而严重影响了光催化产氢效率;同时,浓度猝灭效应也是导致高EY浓度时产氢速率降低的重要原因之一。当EY浓度过高时,激发态的染料物种与另一个基态或激发态的染料物种发生碰撞,并通过非辐射跃迁、辐射跃迁和系间窜跃导致激发态染料的失活30。
3.3.2Pt含量(w)对g-C3N4-NS/Pt光催化产氢活性的影响
图10为Pt含量(w)对g-C3N4-NS/Pt光催化产氢活性的影响。从图中可以看出Pt的最佳含量为1%,产氢速率最大达到282.92 μmol·h-1。随着Pt含量从1%增加到2%、3%和5%时,产氢速率下降。这可能是由于过量的Pt在g-C3N4表面堆积,导致电荷分离受到抑制。
3.3.3EY-g-C3N4-NS/1%Pt体系表观量子效率测定
为了探究光催化产氢与波长的关系,测定了催化剂EY-g-C3N4-NS/1%Pt在不同波长(430、460、490、520、550 nm)下的表观量子效率。在图11中,随着波长的增加,AQEs降低。在430 nm波长下,催化剂EY-g-C3N4-NS/1%Pt的AQE最高(43.6%),这是由于430 nm的光子具有较高的能量。其次是在520 nm,AQE达到23.8%,这主要是因为EY的最大吸收波长在518 nm38。
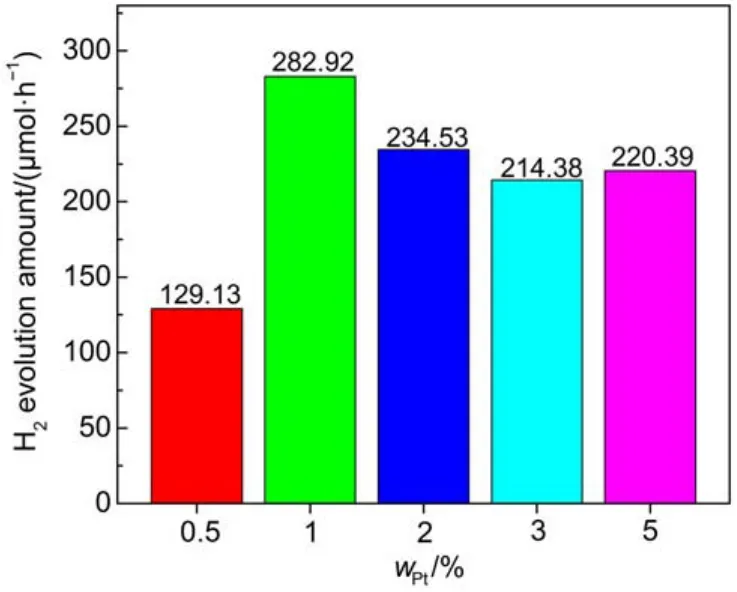
图10 Pt含量(w)对EY-g-C3N4-NS/Pt光催化产氢活性的影响Fig.10 Effect of the different Pt contents(w)on the photocatalytic H2evolution over EY-g-C3N4-NS/Pt

图11 EY-g-C3N4-NS/1%Pt体系在不同波长下的产氢表观量子效率(AQEs)Fig.11 Apparent quantum yields(AQEs)of hydrogen evolution for EY-g-C3N4-NS/1%Pt systems under different wavelength irradiation
3.4EY与石墨烯纳米片g-C3N4-NS之间的电子传递机制
3.4.1荧光光谱分析(PL)
图12为EY敏化g-C3N4和g-C3N4-NS的稳态荧光光谱,通过荧光光谱测试对EY-g-C3N4-NS催化剂的光生电子转移和光催化产氢活性提高的作用机制做了研究。如图所示,EY(1×10-6mol·L-1)在480 nm光激发条件下产生强烈的荧光发射,最大发射峰位于537 nm。当向EY溶液中加入g-C3N4之后,其荧光强度发生轻微下降;当g-C3N4-NS存在时,其荧光强度明显下降,其原因是由于g-C3N4-NS相比于体相g-C3N4具有更高的比表面积、更大的带隙、更长的光生载流子寿命和更好的电子传导性能,从而显著地提高了光电子的转移效率,使得g-C3N4-NS具有优异的光催化性能。
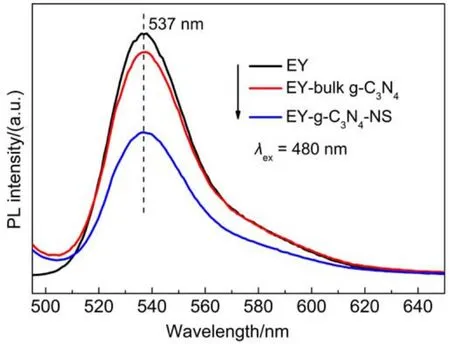
图12 EY敏化(1×10-6mol·L-1)g-C3N4和g-C3N4-NS在10%(φ)TEOA(pH 8)水溶液中的荧光光谱图Fig.12 Fluorescence spectra of EY-sensitized(1×10-6mol·L-1)g-C3N4and g-C3N4-NS samples in 10%(φ) TEOAaqueous solution at pH 8
为了进一步了解激发态EY的电荷转移和其淬灭机制,采用瞬态荧光光谱技术对g-C3N4和g-C3N4-NS存在时激发态EY的荧光淬灭行为进行了表征,拟合得到的数据如表2所示,可以看出,在TEOA溶液中单重激发态EY的荧光衰减可用单指数荧光衰减动力学描述,说明在激发条件下仅存一个发光物种,寿命为0.378 ns。当向EY溶液中加入g-C3N4之后,仍遵循单指数荧光衰减,寿命为0.438 ns,表明g-C3N4存在时单重激发态EY的淬灭方式为静态淬灭机理(static quenching),说明EY吸附于g-C3N4表面形成了具有较弱荧光发射性能的基态复合物(EY-g-C3N4)。当g-C3N4-NS加入后,单重激发态EY的寿命明显增加,其荧光长寿命分别为0.546 ns,且EY的荧光衰减仍用单指数荧光衰减动力学描述,说明在g-C3N4-NS存在时EY的荧光淬灭仍为静态淬灭。该结果表明,g-C3N4-NS存在时可明显延长单重激发态EY的寿命,其优异的电子传输特性以及量子限域效应可显著延长单重激发态EY的寿命,这将有利于EY单重态向三重态的跃迁以及TEOA存在条件下强还原性染料自由基物种EY•-

表2g-C3N4和g-C3N4-NS存在时EY在10%(φ)TEOA水溶液中的荧光寿命Table 2 Fluorescence lifetime of EY in the presence of g-C3N4and g-C3N4-NS in 10%(φ)TEOAaqueous solution
的生成,从而有利于光催化产氢反应的进行。
3.4.2光电化学表征测试
为了进一步证实上述电子迁移的机理,我们对EY敏化体系在TEOA溶液中的光电化学性质进行了测试。图13(A)为EY敏化的g-C3N4和g-C3N4-NS电极的瞬态光电流-时间曲线。从图可以发现,在可见光下氧化铟锡(ITO)/EY-g-C3N4-NS电极的光电流响应能力明显优于ITO/EY-g-C3N4电极。该结果进一步说明EY-g-C3N4-NS界面间的电子迁移效率明显优于g-C3N4,也就是说由于g-C3N4-NS具有量子限域效应可以提高电子转移效率,并且能够增强光激发载流子寿命(相比于体相g-C3N4),从而提高了体系光生电荷的分离效率和光催化分解水产氢的效率。
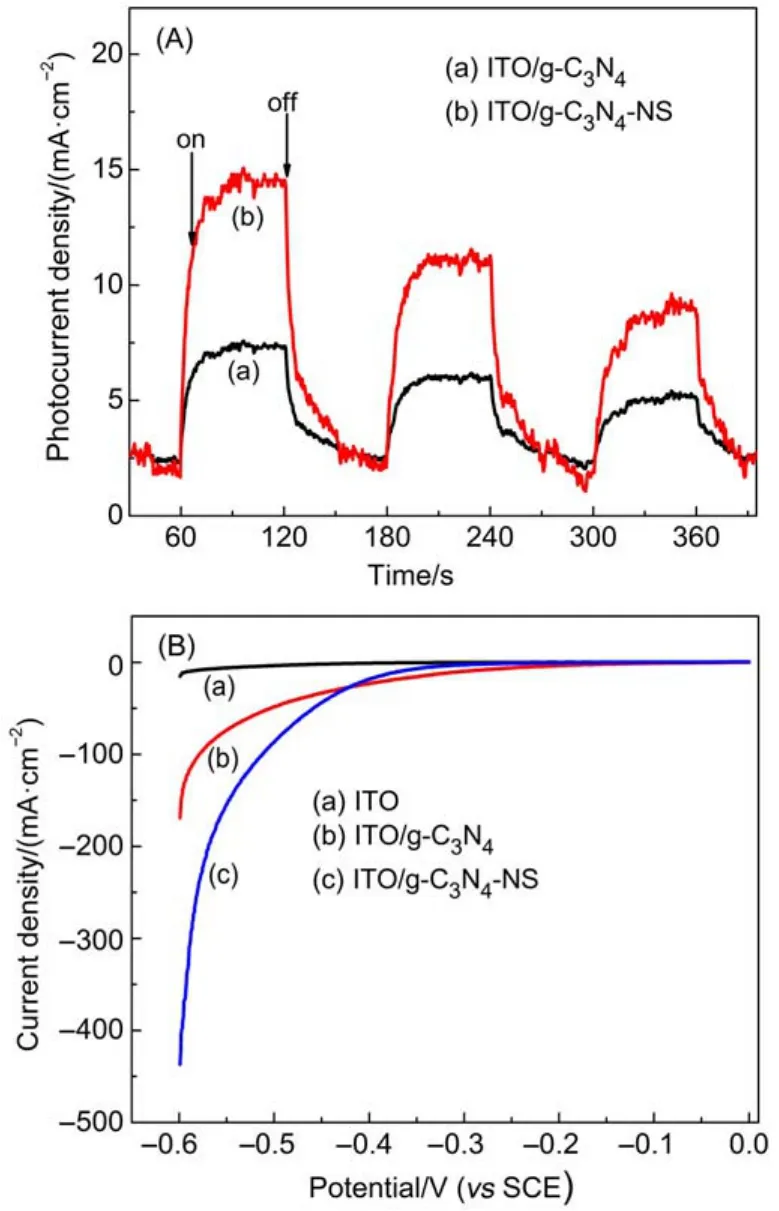
图13 曙红敏化g-C3N4和g-C3N4-NS电极的(A)瞬态光电流-时间曲线及(B)线性扫描伏安(LSV)曲线Fig.13 (A)Transient photocurrent response and (B)linear sweep voltammetry(LSV)curves for the EY-sensitized g-C3N4and g-C3N4-NS electrodes
另外,如前所述,g-C3N4-NS由于其具有量子限域效应可以提高电子转移效率,从而提高了体系光生电荷的分离效率和光催化分解水产氢的效率。为了进一步证实这一点,我们采用线性伏安扫描技术测试了染料敏化g-C3N4-NS材料在TEOA溶液中的电化学分解水产氢活性,并与体相g-C3N4做了比较,结果如图13(B)所示。从图中可以看出,在ITO导电玻璃上可以观察到对应于H+还原为氢气时的阴极电流,且其电流密度在较高的外加电压下依然较低,与之相比,ITO/g-C3N4-NS电极上,在相同的电压范围内还原电流密度明显增加,这表明g-C3N4-NS具有更好的电子传导性能和电子迁移效率,从而提高电催化分解水产氢的活性。该结果说明g-C3N4-NS是一种高效的电催化分解水产氢的催化剂材料。
3.5EY敏化g-C3N4-NS/Pt光催化产氢机理
基于上面的分析和讨论,我们提出了g-C3N4-NS/Pt作为催化剂在曙红敏化体系中的光催化产氢机理,如图14所示。g-C3N4-NS/Pt催化剂光催化产氢活性的显著提高是由于助催化剂Pt和g-C3N4-NS量子限域效应协同增强光生电荷的分离效率和电子转移效率。在可见光照射下,被吸附于g-C3N4-NS表面的EY分子吸收光子的能量形成单重激发态EY1*,随后通过系间跃迁(ISC)形成更为稳定的三重激发态EY3*,当TEOA作为电子给体时,EY3*被还原淬灭生成具有强还原能力的EY-•物种和氧化态TEOA+,EY-•物种的电子传递到负载于g-C3N4-NS表面的助催化剂Pt还原质子H+为H2分子,同时还原态染料分子回到基态;此外g-C3N4-NS也能够还原质子产氢。由于g-C3N4-NS高的比表面积不仅有利于染料吸附以增加染料分子与催化剂的接触面积,而且更有利于EY-•将电子传递给助催化剂Pt的活性点并参与质子的还原反应,这极大的抑制了光生电荷的复合几率并有效地提高了光生电子的转移速率,从而提高了光催化分解水产氢效率。

图14 可见光下EY-g-C3N4-NS/Pt光催化产氢机理Fig.14 Photocatalytic mechanism for hydrogen evolution over EY-sensitized g-C3N4-NS/Pt under visible light irradiation
4 结论
层状g-C3N4这种二维各向异性的纳米甚至亚纳米级的材料具有独特的物理化学性能。通过高温氧化热刻蚀方法将体相g-C3N4剥离成g-C3N4纳米片材料应用于染料敏化可见光产氢,表现出了较体相g-C3N4的高于2.6倍的产氢速率。通过荧光光谱及光电化学表征研究了EY和g-C3N4纳米片之间的电子迁移过程,热剥离后的g-C3N4纳米片具有非常高的比表面积,不仅可以更为有效地吸附染料分子,还因其量子限域效应大大增强了光生电荷的分离效率和电子转移效率,同时,纳米片g-C3N4扩大了其能隙,改善了沿平面方向的电子传输能力以及光生载流子的寿命,从而显著提高g-C3N4纳米片在光敏化体系中的光催化产氢活性。
References
(1)Yang,Y.;Xia,L.F.;Fan,Z.Y.;Chen,W.;Chen,X.P.;Yuan,J.; Shangguan,W.F.J.Mol.Catal.(China)2014,28,182.[杨俞,夏龙飞,范泽云,陈威,陈小平,袁坚,上官文峰.分子催化,2014,28,182.]doi:10.16084/j.cnki.issn1001-3555.2014.02.011
(2) Peng,S.Q.;Ding,M.;Yi,T.;Li,Y.X.J.Mol.Catal.(China) 2014,28,466.[彭绍琴,丁敏,易婷,李越湘.分子催化, 2014,28,466.]
(3) Li,C.L.;Lei,Z.Q.;Wang,Q.Z.;Cao,F.;Wang,F.; Shangguan,W.F.J.Mol.Catal.(China)2015,29,382.[李曹龙,雷自强,王其召,曹菲,王飞,上官文峰.分子催化, 2015,29,382.]doi:10.16084/j.cnki.issn1001-3555.2015.04.012
(4)Wang,X.;Maeda,K.;Thomas,A;Takanabe,K.;Xin,G.; Carlsson,J.M.;Domen,K.;Antonietti,M.Nat.Mater.2009,8, 76.doi:10.1038/nmat2317
(5) Wang,X.;Blechert,S.;Antonietti,M.ACS Catal.2012,2, 1596.doi:10.1021/cs300240x
(6) Chen,X.F.;Jun,Y.S.;Takanabe,K.;Kazuhiko,M.;Kazunari, D.;Fu,X.Z.;Antonietti,M.;Wang,X.Chem.Mater.2009,21, 4093.doi:10.1021/cm902130z
(7)Hao,X.Q.;Jin,Z.L.;Min,S.X.;Lu,G.X.RSC Advances 2016,6,23709.doi:10.1039/C5RA22102A
(9) Wang,X.;Maeda,K.;Chen,X.F.;Takanabe,K.;Kazunari,D.; Hou,Y.D.;Fu,X.Z.;Antonietti,M.J.Am.Chem.Soc.2009, 131,1680.doi:10.1021/ja809307s
(10) Yan,S.C.;Li,Z.S.;Zou,Z.G.Langmuir 2010,26,3894. doi:10.1021/la904023j
(11) Wang,Y.;Di,Y.;Antonietti,M.;Li,H.R.;Chen,X.F.;Wang, X.Chem.Mater.2010,22,5119.doi:10.1021/cm1019102
(12) Chen,X.F.;Zhang,J.S.;Fu,X.Z.;Antonietti,M.;Wang,X. J.Am.Chem.Soc.2009,131,11658.doi:10.1021/ja903923s
(13) Wu,S.Z.Synthesis,Processing and Modification of Graphitic Carbon Nitride with Enhanced PhotocatalyticActivity.Ph.D. Dissertation,South China University of Technology, Guangzhou,2014.[吴思展.类石墨氮化碳(g-C3N4)的合成、加工处理、修饰及其光催化性能的研究[D].广州:华南理工大学,2014.]
(14) Ma,L.;Kang,X.X.;Hu,S.Z.;Wang,F.J.Mol.Catal.(China) 2015,29,359.[马琳,康晓雪,胡绍争,王菲.分子催化, 2015,29,359.]doi:10.16084/j.cnki.issn1001-3555.2015.04.009
(15) Zhang,J.S.;Wang,B.;Wang,X.C.Prog.Chem.2014,26,19. [张金水,王博,王心晨.化学进展,2014,26,19.] doi:10.7536/PC130519
(16) Zhang,J.;Wang,Y.;Jin,J.;Zhang,J.;Lin,Z.;Huang,F.;Yu,J. G.ACS Appl.Mater.Interfaces 2013,5,10317.doi:10.1021/ am403327g
(17) Dong,F.;Zhao,Z.;Xiong,T.;Ni,Z.L.;Zhang,W.D.;Sun,Y. J.;Ho,W.K.ACS Appl.Mater.Interfaces 2013,5,11392. doi:10.1021/am403653a
(18) Ye,C.;Li,J.X.;Li,Z.J.;Li,X.B.;Fan,X.B.;Zhang,L.P.; Chen,B.;Tung,C.H.;Wu,L.Z.ACS Catal.2015,5,6973. doi:10.1021/acscatal.5b02185
(19) Niu,P.;Zhang,L.L.;Liu,G.;Cheng,H.M.Adv.Funct.Mater. 2012,22,4763.doi:10.1002/adfm.201200922
(20) Li,Y.F.;Li,Y.;Li,K.;Yang,Y.;Li,L.J.;Xing,Y.;Song,S.Y.; Jin,R.C.;Li,M.Chem.-Eur.J.2015,21,17739. doi:10.1002/chem.201502945
(21) Cheng,N.Y.;Tian,J.Q.;Liu,Q.;Ge,C.J.;Qusti,A.H.;Asiri, A.M.;Al-Youbi,A.O.;Sun,X.P.ACS Appl.Mater.Interfaces 2013,5,6815.doi:10.1021/am401802r
(22) Cao,S.W.;Yuan,Y.P.;Fang,J.;Shahjamali,M.M.;Boeya,F. Y.C.;Barber,J.;Loo,J.S.C.;Xue,C.Int.J.Hydrog.Energy 2013,38,1258.doi:10.1016/j.ijhydene.2012.10.116
(23) Tonda,S.;Kumar,S.;Kandula,S.;Shanker,V.J.Mater.Chem. A 2014,2,6772.doi:10.1039/C3TA15358D
(24) Schwinghammer,K.;Mesch,M.B.;Duppel,V.;Ziegler,C.; Senker,J.;Lotsch,B.V.J.Am.Chem.Soc.2014,136,1730. doi:10.1021/ja411321s
(25)Wang,Z.S.;Kawauchi,H.;Kashima,T.;Arakawa,H.Coord. Chem.Rev.2004,248,1381.doi:10.1016/j.ccr.2004.03.006
(26) Zhu,K.;Neale,N.R.;Miedaner,A.;Frank,A.J.Nano Lett. 2007,7,69.doi:10.1021/nl062000o
(27) Jin,Z.L.;Zhang,X.J.;Li,Y.X.;Li,S.B.;Lu,G.X.;Catal. Commun.2007,8,1267.doi:10.1016/j.catcom.2006.11.019
(28) Jin,Z.L.;Zhang,X.J.;Li,Y.X.;Li,S.B.;Lu,G.X.J.Mol. Catal.A:Chem.2006,259,275.doi:10.1016/j. molcata.2006.06.035
(29) Liu,X.;Li,Y.X.;Peng,S.Q.;Lai,H.Acta Phys.-Chim.Sin. 2015,31,612.[刘兴,李越湘,彭绍琴,赖华.物理化学学报,2015,31,612.]doi:10.3866/PKU.WHXB201502041
(30) Li,B.;Lü,G.X.Acta Phys.-Chim.Sin.2013,29,1778.[李波,吕功煊.物理化学学报,2013,29,1778.]doi:10.3866/PKU. WHXB201305302
(31)Takanabe,K.;Kamata,K.;Wang,X.;Antonietti,M.;Kubota,J.; Domen,K.Phys.Chem.Chem.Phys.2010,12,13020. doi:10.1039/C0CP00611D
(32) Min,S.X.;Lu,G.X.J.Phys.Chem.C 2012,116,19644. doi:0.1021/jp304022f
(33) Xu,J.J.;Li,Y.X.;Peng,S.Q.;Lu,G.X.;Li,S.B.Phys.Chem. Chem.Phys.2013,15,7657.doi:10.1039/C3CP44687E
(34)Wang,Y.B.;Hong,J.D.;Zhang,W.;Rong,X.Catal.Sci. Technol.2013,3,1703.doi:10.1039/C3CY20836B
(35) Hao,X.Q.;Jin.Z.L.;Wang,F.;Xu,J.;Min,S.X.;Yuan,H.; Lu,G.X.Superlatt.Microstruct.2015,82,599.doi:10.1016/j. spmi.2015.02.028
(36) Hao,X.Q.;Jin,Z.L.;Lu,G.X.Chem.Lett.2016,45,116. doi:10.1246/cl.150886
(37) Ge,L.;Han,C.;Xiao,X.;Guo,L.Int.J.Hydrog.Energy 2013, 38,6960.doi:10.1016/j.ijhydene.2013.04.006
(38) Zhen,W.L.;Ma,J.T.;Lu,G.X.Appl.Catal.B-Environ.2016, 190,12.doi:10.1016/j.apcatb.2016.02.061
(39)Williams,G.;Kamat,P.V.Langmuir 2009,25,13869. doi:10.1021/la900905h
Quantum Confinement Effect of Graphene-Like C3N4Nanosheets for Efficient Photocatalytic Hydrogen Production from Water Splitting
HAO Xu-Qiang1YANG Hao1JINZhi-Liang1,*XU Jing1MINShi-Xiong1LÜ Gong-Xuan2,*
(1School of Chemistry and Chemical Engineering,Beifang University of Nationalities,Yinchuan 750021,P.R.China;2State Key Laboratory for Oxo Synthesis and Selective Oxidation,Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences,Lanzhou 730000,P.R.China)
Nanosheet materials obtained from laminar compounds are new two-dimensional anisotropic nanomaterials that can even reach the sub-nanometer scale.These materials possess unique physical and chemical properties.An example of such a nanosheet materials is graphitic carbon nitride(g-C3N4)nanosheets transformed from bulk g-C3N4.Here,g-C3N4nanosheets were prepared from bulk g-C3N4by high-temperature thermal oxidation.The photocatalytic activity of eosin(EY)-sensitized g-C3N4nanosheets for hydrogen evolution was about 2.6 times higher than that of bulk g-C3N4.The structure of the g-C3N4nanosheets and process of electron transfer between EY and the g-C3N4nanosheets were investigated by X-ray diffraction(XRD),Fourier transform infrared(FTIR)spectroscopy,scanning electron microscopy(SEM),Brunauer-Emmett-Teller(BET) analysis,fluorescence spectroscopy,and photoelectrochemical measurements.The g-C3N4nanosheets possessed high specific surface area.The g-C3N4nanosheets not only effectively absorbed dye molecules,but also enhanced the separation and electron transport efficiencies of photogenerated charges because of their quantum confinement effect.The quantum confinement effect of g-C3N4nanosheets widened their bandgap, improved electron transfer ability along the in-plane direction,and lengthened the lifetime of photoexcited charge carriers.As a result,the photocatalytic activity of the g-C3N4nanosheets was improved compared with that of bulk g-C3N4.
April 21,2016;Revised:June 21,2016;Published online:June 22,2016.
s.JIN Zhi-Liang,Email:zl-jin@nun.edu.cn;Tel:+86-951-2067915.LÜ Gong-Xuan,Email:gx-lu@lzb.ac.cn; Tel:+86-931-4968178.
g-C3N4nanosheet;Dye-sensitization;Quantum confinement effect;Photocatalysis; Hydrogen evolution
O643
10.3866/PKU.WHXB201606226
The project was supported by the National Natural Science Foundation of China(21263001,21463001,21433007).
国家自然科学基金(21263001,21463001,21433007)资助项目©Editorial office ofActa Physico-Chimica Sinica
(8) Cao,S.;Yu,J.J.Phys.Chem.Lett.2014,5,2101.10.1021/ jz500546b
猜你喜欢
科学之友(2022年11期)2022-11-03
工业水处理(2022年6期)2022-06-23
世界科学技术-中医药现代化(2022年2期)2022-05-25
世界科学技术-中医药现代化(2021年8期)2021-12-21
世界科学技术-中医药现代化(2021年7期)2021-11-04
石油化工高等学校学报(2021年3期)2021-07-15
陶瓷学报(2019年5期)2019-01-12
无机盐工业(2017年5期)2017-05-25
郑州大学学报(理学版)(2017年1期)2017-04-07
物理化学学报(2017年3期)2017-03-11
